

介绍DS-PAW aimd分子动力学模拟的内容
描述
第一性原理平面波密度泛函计算软件DS-PAW是Device Studio平台下的一款使用C++开发的国产第一性原理密度泛函计算软件,使用平面波作为基函数组,其赝势是使用投影缀加平面波方法构造的。
DS-PAW能够应用于不同场景,例如金属、半导体、绝缘体、表面、磁性、非磁性和锂电等;能够精确预测材料的电子分布;能够进行原子几何结构优化;能够广泛的应用于材料科学领域。
本期将给大家介绍DS-PAW aimd分子动力学模拟的内容。
2.18. aimd分子动力学模拟
本节将以水分子体系为例,介绍在DS-PAW中如何进行分子动力学模拟计算。
2.18.1. H₂O分子动力学模拟输入文件
输入文件包含参数文件 aimd.in 和结构文件 structure.as , aimd.in 如下:
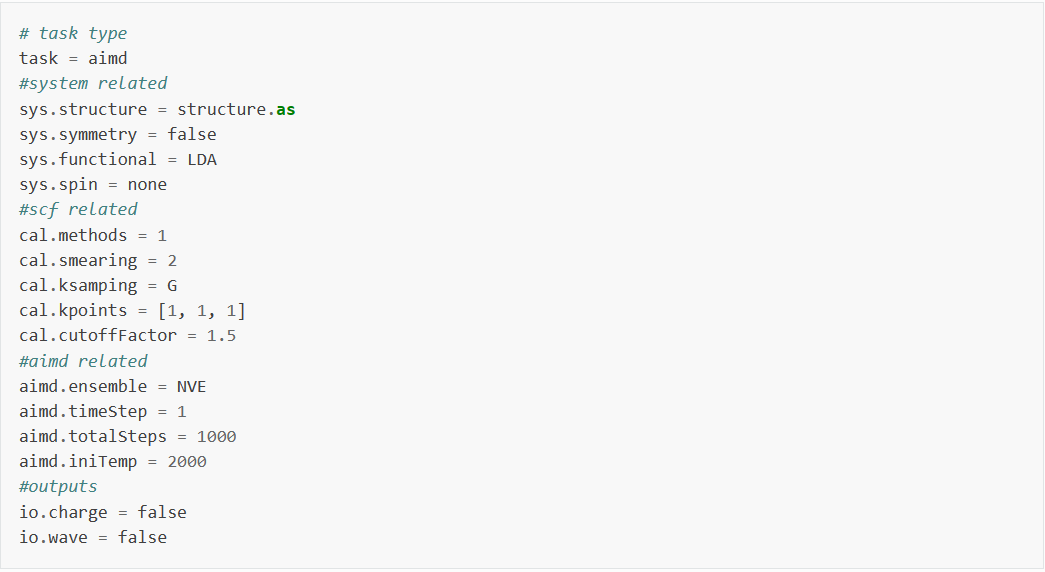
aimd.in 输入参数介绍:
在分子动力学模拟计算中可以尽量保留sys.和cal.的参数到 aimd.in 中,之后设置分子动力学模拟计算特有的参数即可:
task : 本次计算为分子动力学计算,设置task为aimd;
aimd.ensemble : 表示分子动力学模拟时选用的系综;
aimd.timeStep : 表示分子动力学模拟时的时间步长;
aimd.totalSteps : 表示分子动力学模拟的总步数;
aimd.iniTemp : 表示分子动力学模拟时的初始温度;
structure.as 文件参考如下:

2.18.2. run程序运行
准备好输入文件之后,将 aimd.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW aimd.in 。
2.18.3. analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 aimd.json 、 paw_tmp/aimd.tmp 这3个文件。
aimd.json : 分子动力学计算完成之后的 json 数据文件;此时模拟时间内原子位置、体系能量和温度等数据被保存在 aimd.json 中,具体的数据结构详见数据结构解析部分;
paw_tmp/aimd.tmp : 分子动力学计算中的轨迹文件,默认aimd计算中每20个离子步记录一次结构信息;
使用 Device Studio 可直接对 aimd.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 aimd.json 即可,可根据作图要求自定义设置面板参数。
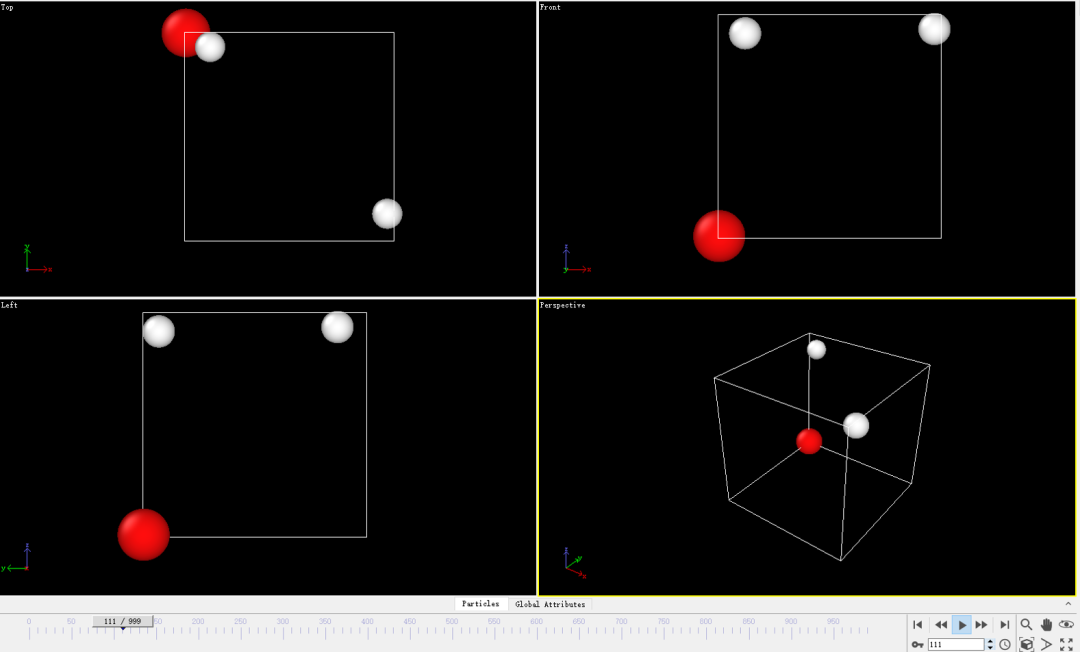
DS可以播放在模拟过程中分子的运动轨迹,截取其中一帧的结构图如下图所示:
另可使用 python 进行数据处理,将 aimd.json 转为 pdb 文件,具体操作见 辅助工具使用教程 部分。

审核编辑:刘清
-
介绍DS-PAW efield外加电场计算的内容2023-02-22 2796
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 510
-
热分析动力学2009-12-01 719
-
拓展人类潜能:深势科技使用IPU赋能分子动力学2021-12-09 3279
-
Device Studio应用实例之DS-PAW2022-08-10 4023
-
DS-PAW应用实例2022-08-11 3556
-
GORMACS如何使用?一个方法快速完成动力学模拟计算2022-11-14 3755
-
DS-PAW scf自洽计算的内容2023-01-31 2773
-
DS-PAW band能带计算2023-02-02 2640
-
DS-PAW bandunfolding能带反折叠计算2023-02-25 3094
-
DS-PAW bader电荷计算2023-02-27 3719
-
NTO表面和分子空位对其热反应动力学的影响2023-05-19 4129
-
使用分子动力学轨迹来预测大型自旋系统中的核自旋弛豫行为2023-05-20 2361
-
基于车辆动力学模型的横向控制2023-11-15 2092
-
SCI 期刊验证!苏黎世大学使用 ALINX FPGA 开发板实现分子动力学模拟新方案2025-09-22 761
全部0条评论

快来发表一下你的评论吧 !
