

基于LiPF6开发的新型低温电池电解液
描述
研究背景
基于LiPF6的商用电解质广泛用于锂离子电池 (LIB)。然而,由于EC的凝固点高导致离子电导率低,脱溶剂过程缓慢,界面电阻大,目前商用的基于LiPF6的电解质的锂离子电池在低温运行中表现不尽如人意。在LiPF6基于LHCEs,LiPF6总是形成界面阻抗高的SEI膜,不能满足低温应用的要求,因此成膜添加剂是非常必要的。常用的添加剂包括氟碳酸乙烯酯(FEC)、二氟磷酸锂(LiPO2F2)等,可以形成界面阻抗低的富LiF的SEI膜,以提高Li在低温下的扩散。
成果简介
近日,中科院陈成猛研究员、徐国宁研究员开发了一种基于LiPF6的局部高浓度电解质,与二氟双(草酸)磷酸锂(LiDFBOP)添加剂协同提高LIB在低温下的性能。在不含 EC溶剂中溶解较高浓度的 LiPF6提高了电解质的整体离子电导率。且Li+–溶剂–PF6–的结构有利于去溶剂化能的降低;同时,引入LiDFBOP构建具有高离子电导率和循环稳定性的SEI膜。实现石墨/锂电池良好的倍率性能和低温性能(-20 °C(0.1 C)时约 240 mAh g–1)。这项工作为开发商用LiPF6基电解质以改善 LIB 在低温下的运行提供了可行的策略。该工作以“Boosting the Low-Temperature Performance for Li-Ion Batteries in LiPF6-Based Local High-Concentration Electrolyte”为题发表在ACS Energy Letters上。
研究亮点
(1) 提出一种基于LiPF6的局部高浓度电解质用于提高 LIB 在低温下的性能。 (2) 在不含EC溶剂中较高浓度的 LiPF6提高了电解质的整体离子电导率 (3) Li+–溶剂–PF6–的结构有利于降低脱溶剂化能。 (4) 石墨/锂电池在低温下-20 °C(0.1 C)的容量约 240 mAh g–1。
图文导读
高浓度电解质(HCE)为2 M LiPF6-DMC。通过在HCE中添加LiDFBOP来制备含LiDFBOP的高浓度电解质(HCE-P)。将1,1,2,2-四氟乙基-2,2,3,3-四氟丙醚(HFE)作为电解质的稀释剂加入上述高浓度电解液中;两个LHCE分别被命名为LHCE和LHCE-P。
此外,商用电解质(1 M LiPF6在 EC:DMC:DEC 中,体积比为 1:1:1)用作参比电解液 (RCE)。五种电解质、DMC 和 HFE 的拉曼光谱如图 1a 所示。可以看到,在HCE中,属于DMC峰值为915cm–1移至 923 和 939 cm–1,且游离DMC减少甚至消失,转化为配位DMC。同时,代表配位PF 6–的峰出现在748 cm–1,这与游离PF 6–的峰值不同(在RCE中716和741cm–1)。这表明在HCE中形成独特的溶剂化结构。
值得注意的是,含HFE稀释剂的LHCE拉曼光谱峰与HCE几乎相同,除了出现在825–875 cm–1左右的峰起源于HFE。这证实了引入HFE后可以保持HCE的溶剂化结构。在HCE-P和LHCE-P中也可以找到类似的结果。因此,LiDFBOP的添加不会破坏LHCE中独特的溶剂化结构。
在图 1b 中,7Li NMR结果表明,高浓度电解质(HCE和HCE-P)的化学位移高于低浓度电解质(RCE),表明Li周围的溶剂化作用增强。此外,尽管加入HFE后,LHCE和LHCE-P的化学位移向较低的场,但它们仍然高于RCE,表明Li+和PF6–之间的相互作用更强。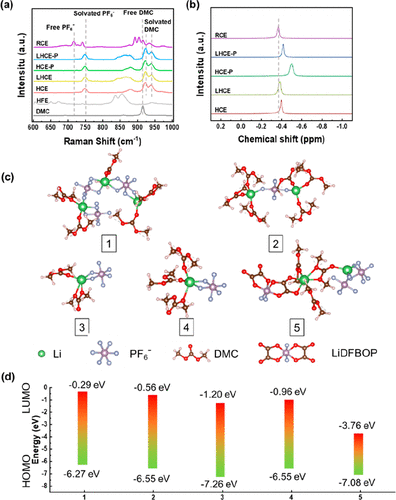
图1.(a) DMC、HFE、HCE、LHCE、HCE-P、LHCE-P 和 RCE 的拉曼光谱。(b)7在25°C室温下五种电解质的Li NMR结果。(c)由AIMD模拟结果得到五种溶剂化配合物。(d)计算c部分中溶剂化配合物的HOMO/LUMO能。 为了验证独特溶剂化结构的形成,HCE,HCE-P的分子动力学(AIMD模拟结果,图1c)显示出5种配位体构型及相应的脱溶剂化能(表1),Li+-4 DMC的脱溶能为6.00eV,高于其他络合物的脱溶能。
因此,特殊的Li+-solvent-PF6-结构可能通过促进脱溶过程的动力学来改善速率性能和低温性能,这将在后面讨论。最后,为了验证LiDFBOP添加剂的作用,使用DFT计算了这些配合物的最高占有分子轨道(HOMO)和LUMO能级。在图1d中,配合物5的LUMO能级比配合物1、2、3和4的能级低很多。
换句话说,LiDFBOP的存在会降低配合物中LUMO的能级,使其容易被电子填充。因此,我们推测LiDFBOP会优先反应,在HCE-P和LHCE-P中生成SEI膜。
表1 不同配位结构的脱溶剂化能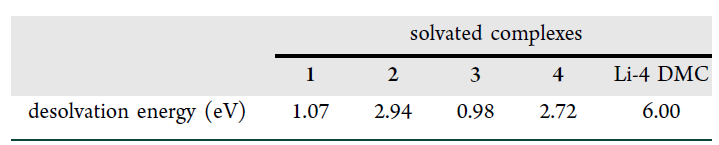
图2a显示了石墨/Li半电池的第一个循环CV曲线。RCE的CV曲线在0.7 V附近显示一个峰值,对应于EC的分解和SEI膜的形成。在HCE和LHCE中,由于PF6–被挤压到Li溶剂化内层中,PF6–在0.4 V分解以产生的 SEI膜。在HCE-P和LHCE-P中,锂插入过程中仅出现一个2 V左右的峰,实验证实LiDFBOP将优先于EC和LiPF6分解。.由图2b中的第一条充放电曲线可以看出,LiDFBOP的这种优先还原将防止锂盐和溶剂的分解,从而提高初始库仑效率(ICE)。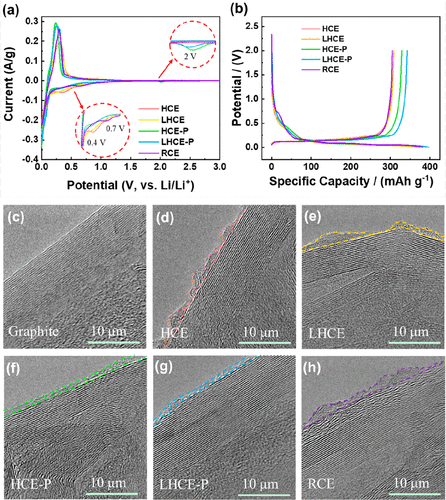
图2.(a)五种电解质中石墨/锂电池的第一周期CV曲线和(b)初始充放电曲线。(c) 石墨原料透射电镜。不同电解质中SEI膜的TEM:分别为HCE(d),LHCE(e),HCE-P(f),LHCE-P(g)和RCE(h)
使用透射电子显微镜(TEM)观察SEI膜的微观结构。作为比较,原始石墨显示出如图2c所示的光滑表面。活化后,在HCE,LHCE和RCE中形成的SEI膜的厚度不均匀(图2d,e和h)。这很可能是因为由EC和LiPF6分解形成的SEI膜表现出较差的结构稳定性,导致初始SEI膜随着石墨体积的增加而破裂。结果,更多的锂盐和溶剂被不断消耗以形成更厚的SEI膜。
在含有添加剂的电解质中,LiDFBOP有助于形成具有更高机械强度的SEI膜,从而减缓SEI膜破裂的可能性。因此,SEI膜在HCE-P和LHCE-P中变得相对均匀和薄(图2f,g)。
图3.在LHCE,LHCE-P和RCE电解质中循环的石墨阳极表面上C 1s(a)和F 1s(c)的XPS光谱。不同Ar溅射时间下石墨阳极表面C 1s(b)和F 1s(d)光谱的XPS结果。
通过图3中的X射线光电子能谱(XPS)检测在不同电解质中形成的SEI膜的化学成分。C-C/C-H峰(284.8 eV)代表被SEI膜覆盖的石墨,可以间接反映SEI膜的厚度。在图3a中,含有LiDFBOP(HCE-P,LHCE-P)的电解质中的C-C/C-H峰远高于不含LiDFBOP(HCE,LHCE)的电解质,这意味着LiDFBOP有利于形成更薄的SEI膜,如图2f和g所示。
一般来说,LiF由于其高杨氏模量而对SEI膜做出了积极贡献。不幸的是,在RCE中检测到较少的LiF,这意味着SEI膜可能不够稳定,无法承受石墨体积的变化,导致SEI膜破裂。在LHCE中,SEI含有大量的LiF,有机成分很少,可以提供足够的灵活性来适应石墨体积的变化。
在HCE-P和LHCE-P中,LiF,C-O,C═O(Li2C2O4) 和LixPOyFz(LiDFBOP的分解产物)与不含LiDFBOP的电解质相比均增加,如图3a和c部分所示,这赋予SEI膜高离子电导率和优异的稳定性。
通过进行6、12和18 s的深度Ar溅射进一步分析SEI膜的深度剖面。在HCE和LHCE(图3b和d)中,SEI膜中LiF,C-O和C═O的含量没有明显的变化,这意味着SEI膜呈现有机和无机混合随机分布的马赛克结构。这些组分的不均匀分布可能导致不同的离子在运输过程中克服不同的能量势垒,使离子传输更加困难。
此外,这种结构加剧了电解质和电子的还原,消耗了可逆的锂离子。在HCE-P和LHCE-P(图3b和d)中,随着溅射时间的增加,C-O、C═O和LixPOyFz呈下降趋势,而LiF,Li2CO3, and LixPFy呈上升趋势。这些结果表明,LiDFBOP衍生的SEI膜表现出经典的多层结构。其内层主要由无机物质组成,外层则由有机聚合物组成。
众所周知,SEI膜中的无机成分可以提供高导锂性,而有机部分提供了缓冲体积变化的高弹性,保持了界面层的完整性。这种致密的SEI膜可以通过防止电解质与石墨直接接触来抑制电解质的分解,从而获得更好的循环性能。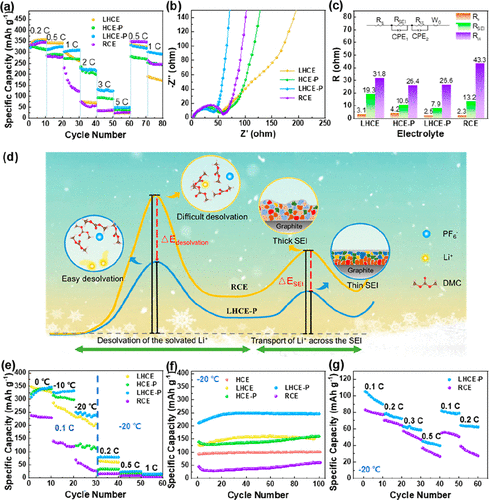
图4.(a)石墨/锂电池的倍率性能,(b)第3圈的EIS以及(c)室温下使用四种电解质的石墨/锂电池的相应阻抗值。(d)LHCE-P改进动力学的示意图。(e)不同电流密度下的比容量和(f)使用五种电解质的石墨/锂电池在低温下的循环性能。(g)使用LHCE-P 和 RCE 的 NCM622/石墨软包电池在 −20 °C 下的倍率性能。
最后,作者测试了含有四种电解质的石墨/锂电池在25 °C下的电化学性能。由图4a所示,添加了HFE的LHCE-P的倍率能始终优于HCE-P。这主要是由于LHCE和HCE之间的差异。虽然两者都显示出高Li的迁移率,但LHCEs中的稀释剂可以有效降低电解质的粘度,提高电解质对电极材料的润湿性。石墨/锂电池在充放电3个圈后的EIS可以验证中的这一推论图4b,c。通常,电池的EIS由欧姆阻抗(Rs)、SEI膜的阻抗(RSEI)和电荷转移电阻(Rct)。HCE-P 显示最高的Rs,可能是因为高粘度和低润湿性增加了电池的固有阻抗。
在图4a中,与RCE和LHCE相比,添加了LiDFBOP的电解质显示出更高的比容量。例如,在 2 C 下HCE-P和LHCE-P的容量约为210 mAh g–1。这可能是因为有利的化学成分和结构大大降低了Li通过SEI膜的阻抗。具体而言,正如TEM和XPS所证实的那样,LiDFBOP衍生的SEI更薄,从而减少了Li的传输路径。此外,在SEI膜中更多的LiF和Lin(FOP)n促进Li的扩散。利用EIS验证了LiDFBOP可以降低Li通过SEI膜的阻抗。在图4 b 和 c中,使用LiDFBOP电解质的RSEI总是低于没有LiDFBOP的电解质。
更重要的是,在图4a中,当电流密度在5 C下进行高倍率测试后回到1 C时,HCE-P、LHCE-P和RCE中石墨/锂电池的比容分别量可以完全恢复到300、250和253 mAh g–1。而LHCE的容量无法恢复到以前的水平。正如我们前面提到的,LiPF6-LHCE中衍生的SEI膜含有较少的有机物质,无法适应石墨体积的变化,导致SEI膜的持续生长和可逆容量的降低。因此,可逆容量的降低归因于SEI膜的重建和失效。与HCE-P 和 LHCE-P的Rs和RSEI相比,在没有LiDFBOP的电解质中R s和RSEI大大增加。
众所周知,中频阻抗与Li脱溶剂的过程有关。Li–solvent–PF6–的Rct的值含(HCE-P、LHCE、LHCE-P)始终低于RCE。因此,实验证实这种独特的Li–solvent–PF6–溶剂化结构有利于Li的脱溶剂化过程,这与表1的理论结果相当吻合。 为了验证电解液的实用性,进一步测试了锂||石墨电池的低温性能。图4d进一步计算了Li嵌入石墨过程中每一步的能垒:首先,LHCE-P中的局部高浓度带来了相对较低的Li迁移能垒;其次,得益于Li–solvent–PF6–结构,Li脱溶剂能降低,实现Li快速脱溶剂动力学。第三,LHCE-P中LiDFBOP衍生的SEI膜表现出更高的离子电导率,促进了Li在低温下通过SEI膜的迁移。
因此,在0.1C的速率下和-10°C温度下,LHCE-P可以将其性能保持在320 mAh g–1左右(图4e)。即使在−20°C下,LHCE-P仍表现出240 mAh g–1。相比之下,由于RCE中的EC表现出较高的熔点,并且随着温度的降低,Li脱溶剂障碍进一步加剧,因此在低温下始终表现出最低的性能。
随后,分别在-20°C下以0.2、0.5 C和1C的倍率测试电池的充放电性能。为了确保电解液在低温下能够继续充放电,进一步测试了电池在−20°C的低温循环。如图4f所示,尽管RCE在循环期间显示出比容量的小幅增加,但总体上总是显示低容量。
相比之下,LHCE中的Li–solvent–PF6–结构可以降低脱溶剂能,表现出比RCE更高的容量,但LiPF6-分解形成的SEI膜机械强度较低,导致循环过程中的大量容量损失。添加LiDFBOP添加剂后,LiF和Lin(FOP)n多孔有机层促进Li的扩散,而Li+2C2O4和LiF使SEI膜更稳定。该添加剂还有助于形成更薄的SEI,从而减少Li的运输路径。得益于优异的SEI膜和独特的溶剂化结构,LHCE-P具有良好的容量和良好的循环性能。
与HCE相比,HCE-P性能的提高再次证明了LiDFBOP的作用(图4f)。然而由于它们的高粘度,对电极的润湿性差,以及电解质的阻抗大,这两种高浓度电解质(HCE和HCE-P)与局部高浓度电解质(LHCE和LHCE-P)相比表现出较差的性能。 作者进一步测试了LiNi0.6Co0.2Mn0.2O2(NCM622)/石墨软包电池在-20°C下的性能。如图4g所示,LHCE-P在比容量和容量保持方面始终优于RCE。在 0.1 C,LHCE-P的比容量达到108 mAh g–1,高于 RCE(80 mAh g–1)。随着循环次数的增加,虽然两种电解质的可逆容量丧失,但RCE的可逆容量明显迅速下降。
总结与展望
作者通过设计了一种基于LiPF6的局部高浓度电解液,并以LiDFBOP作为牺牲性添加剂,以改善LIB的低温性能。LHCE的应用有效地提高了Li的导电性;同时,LiDFBOP的引入可通过生成具有低相间阻抗的稳定SEI膜,保证了LiPF6基LHCE的应用。受益于电极-电解质界面化学的稳定性,商用石墨负极表现出良好的倍率性能(在2C时约为225 mAh g-1)和令人印象深刻的低温性能(在-20℃(0.1C)时约为240 mAh g-1)。更重要的是,这项工作证明了Li–solvent–PF6–结构可以降低脱溶剂化能,从而为开发功能性电解质提供了可行的策略,以在现有系统的基础上满足实际生活中低温操作的要求。
审核编辑:刘清
-
锂电池电解液如何影响电池质量?锂电池电解液成分优势是什么?2024-01-11 3059
-
电解液与SEI的关系?电解液对SEI的影响?2023-11-10 1907
-
电解液宽温性能的影响因素和宽温电解液研究进展2020-10-21 12986
-
锂电池电解液的组成部分_锂电池电解液的危害2020-08-03 12343
-
锂电池电解液是什么_锂电池电解液主要成分2020-03-30 50996
-
电解液——锂电池的‘血液’2018-08-07 6106
-
锂离子电池电解液超全面介绍 有何神秘之处?2017-02-22 7302
-
锂离子电池电解液有机溶剂的发展趋势2013-06-17 5950
-
锂离子电池材料之电解液(详细篇)2009-11-03 20275
-
锂离子电池电解液研究进展2009-10-30 1740
-
锂离子电池电解液是什么?2009-10-27 14813
-
现在的锂电池都是用什么样的电解液?电解液里加入什么添加剂?2009-10-23 5247
全部0条评论

快来发表一下你的评论吧 !
