

AEM综述:硫化物基固态锂电池的先进表征技术
描述
01
导读
硫化物固体电解质具有非流动性、单离子导电性和低可燃性等优点,有望实现高能量和功率密度的下一代二次电池。然而,固体电解质的(电)化学分解、界面处的机械降解、锂枝晶生长以及活性材料中锂扩散缓慢等问题限制了其实际应用。
02
成果简介
近日,Advanced Energy Materials上发表了一篇题为“Advanced Characterization Techniques for Sulfide-Based Solid-State Lithium Batteries”的综述,对硫化固体电解质固态锂电池的上述四个问题进行了综述,还介绍了X射线光电子能谱、飞行时间二次离子质谱、透射电子显微镜和X射线计算机断层扫描等先进表征技术在解决这些问题方面的现状和前景。
03
关键创新
概述了硫化物基固态锂电池的主要挑战,以及硫化物基固态锂电池先进表征技术的现状和未来展望。
04
核心内容解读
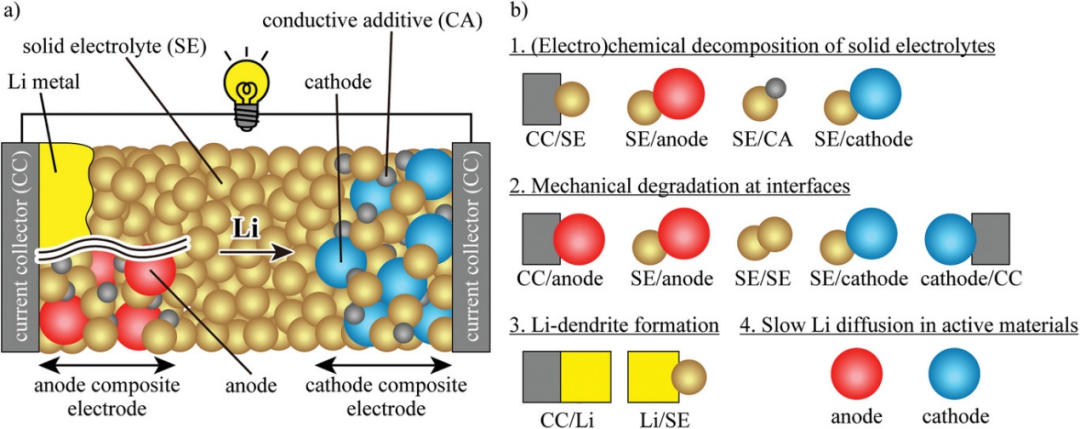
图1. a)固态锂电池构型。b)界面和活性物质中的主要问题。@Wiley
1、硫化物基固态锂电池的问题
图1a显示了含硫化物SEs的固态锂电池示意图。负极是锂金属或复合电极结构。其中,存在各种固-固界面。这篇综述主要关注四个根本问题,即硫化物SEs的(电)化学分解,界面的机械退化,Li枝晶形成,,活性物质中Li扩散缓慢(图1b)。
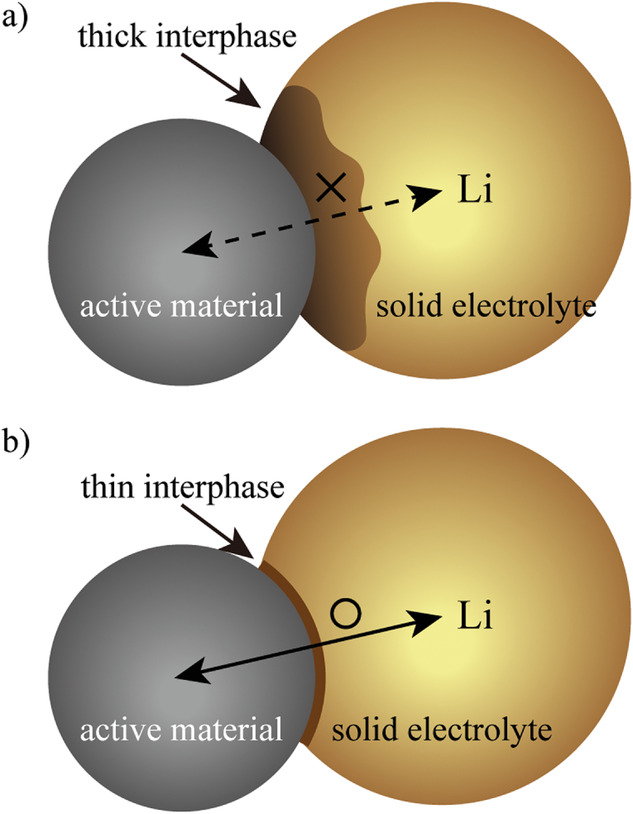
图2. 活性物质和固体电解质(SEs)之间形成的界面示意图。a)高电子导电性界面相。b)低电子导电性界面相。@Wiley
2、硫化物SEs的(电)化学不稳定性
在硫化物SEs中,(电)化学不稳定性是一个重要问题。在高于氧化稳定性的电位下,锂从硫化物SEs中脱出,形成贫锂的正极-电解质界面相(CEI)。在低于还原稳定性的电位下,随着P5+或金属元素的还原,Li被插入到硫化物SEs中,形成富Li的固体电解质界面相(SEI)。
界面相的电子电导率和离子电导率是决定界面电阻的关键。图2a,b显示,当界面相电子电导率较高时,分解后的界面相具有与活性物质相同的电势,导致SE逐渐分解,界面相迅速增厚,阻碍了界面处的Li传输,降低了电池容量。相反,如果界面相是电子绝缘的,在活性物质的电位下,界面相将硫化物SE钝化,从而抑制分解。
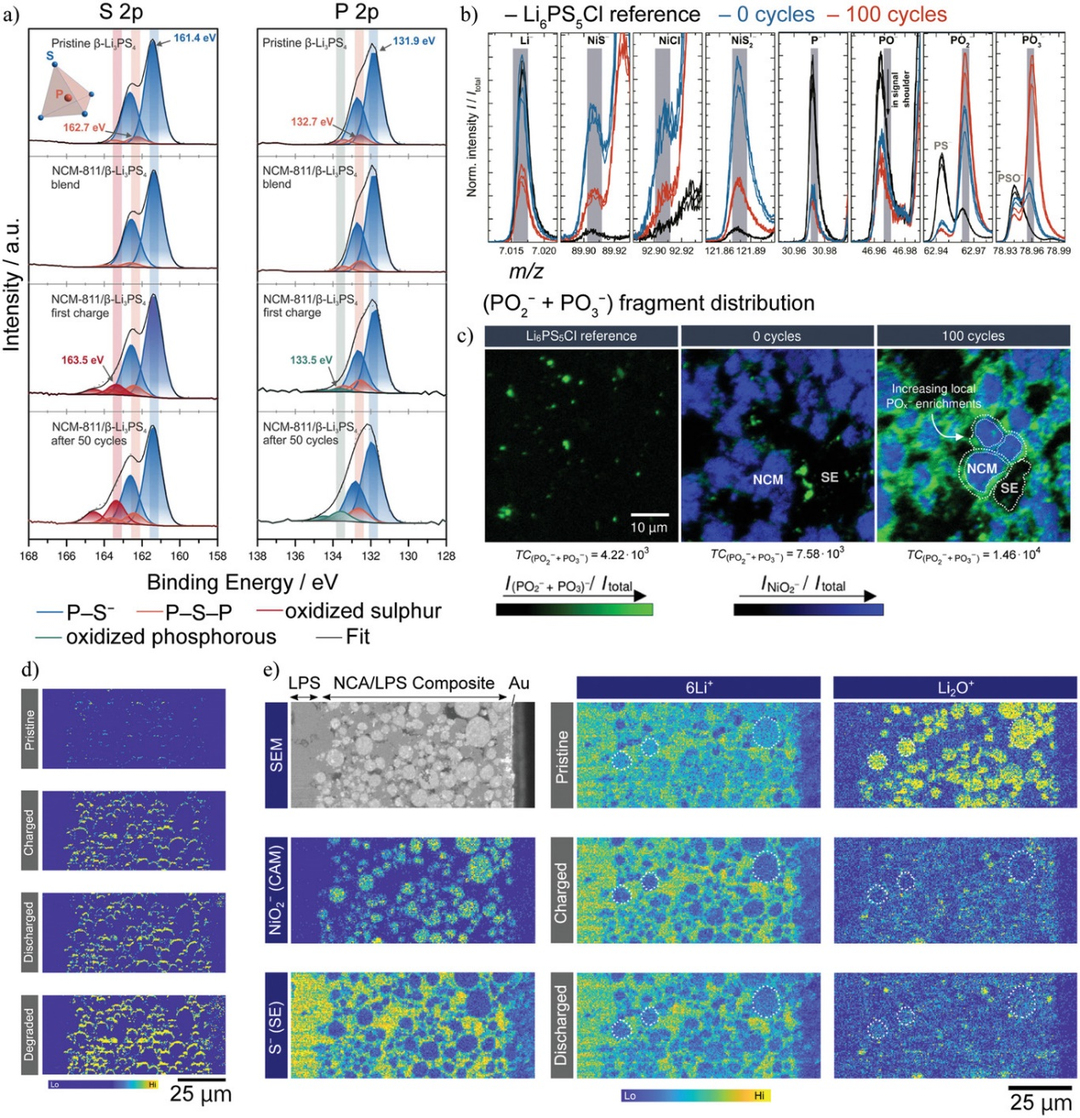
图3. a)β-Li3PS4、LiNi0.8Co0.1Mn0.1O2(NCM-811)/β-Li3PS4复合正极初始状态、第一次充电(0.1C)后、50次循环后的S-2p和P-2p XPS光谱。b)LiNi0.6Co0.2Mn0.2O2/Li6PS5Cl中带负电的离子碎片的飞行时间二次离子质谱(ToF-SIMS)。c)NiO2−(蓝色)和POx−(绿色)碎片的局部分布。d)通过原位ToF-SIMS测量LiNi0.80Co0.15Al0.05O2(NCA)/Li2S-P2S5微晶玻璃(LPS)复合正极中POx−碎片的演化。e)左:NCA/LPS复合正极的扫描电镜(SEM)图像和NiO2−和S−碎片的分布。中:6Li+碎片在充放电循环中分布的演变。右:Li2O+碎片在充放电循环中分布的演变。@Wiley
3、硫化物SEs的CEIs表征
通过XPS可以研究CEI的电子状态。原始状态下,只检测到P-S−(161.4 eV)和P-S-P(162.7 eV)的XPS峰。在充电和循环状态下,在高能量侧观察到一个宽峰,表明CEI含有带有-S-S-键的多硫化物。该宽峰强度随充放电反应而增大或减小,表明CEI随电势变化而发生氧化还原反应。XPS被广泛用于表征界面反应,因为它可以测量SEs和CEIs中S和P电子状态的微小差异。然而,XPS的空间分辨率较低,不能用于研究复合电极中CEIs的空间分布。
还可以使用亚微米空间分辨率的ToF-SIMS表征CEI。与XPS相比,ToF-SIMS的优点是具有更高的空间分辨率和化合物灵敏度。图3b显示了Li6PS5Cl、NCM-622/Li6PS5Cl电极在混合后以及100次循环后的碎片强度。100次循环后,样品中的PO2−和PO3−碎片增加。同样,观察到SO2−和SO3−碎片增加。此外,利用ToF-SIMS的高空间分辨率,还能显示正极附近CEI的空间分布。图3c显示,100次循环后,样品中POx−碎片的强度在NCM-622附近增加,表明硫化物SE与NCM-622发生了反应。
NCA和LPS复合电极中PO2−和PO3−碎片的综合强度分布结果表明,充放电后正极颗粒周围的POx-碎片明显增多。6Li+碎片代表NCA和LPS中的Li分布,而Li2O+碎片代表NCA中的Li分布。6Li+和Li2O+碎片强度在充电时下降,在放电时略有增加。然而,NCA中Li的浓度与碎片强度并没有线性关系。尽管初始充放电效率为80%,但放电状态下6Li+和Li2O+碎片的强度明显低于原始状态。层状岩盐正极的电子电导率在初始Li脱出过程中会增加,这可能会改变碎片的电离概率。另外,表面损伤也会导致放电状态强度低。因此,定量分析显然是ToF-SIMS中的一个问题。
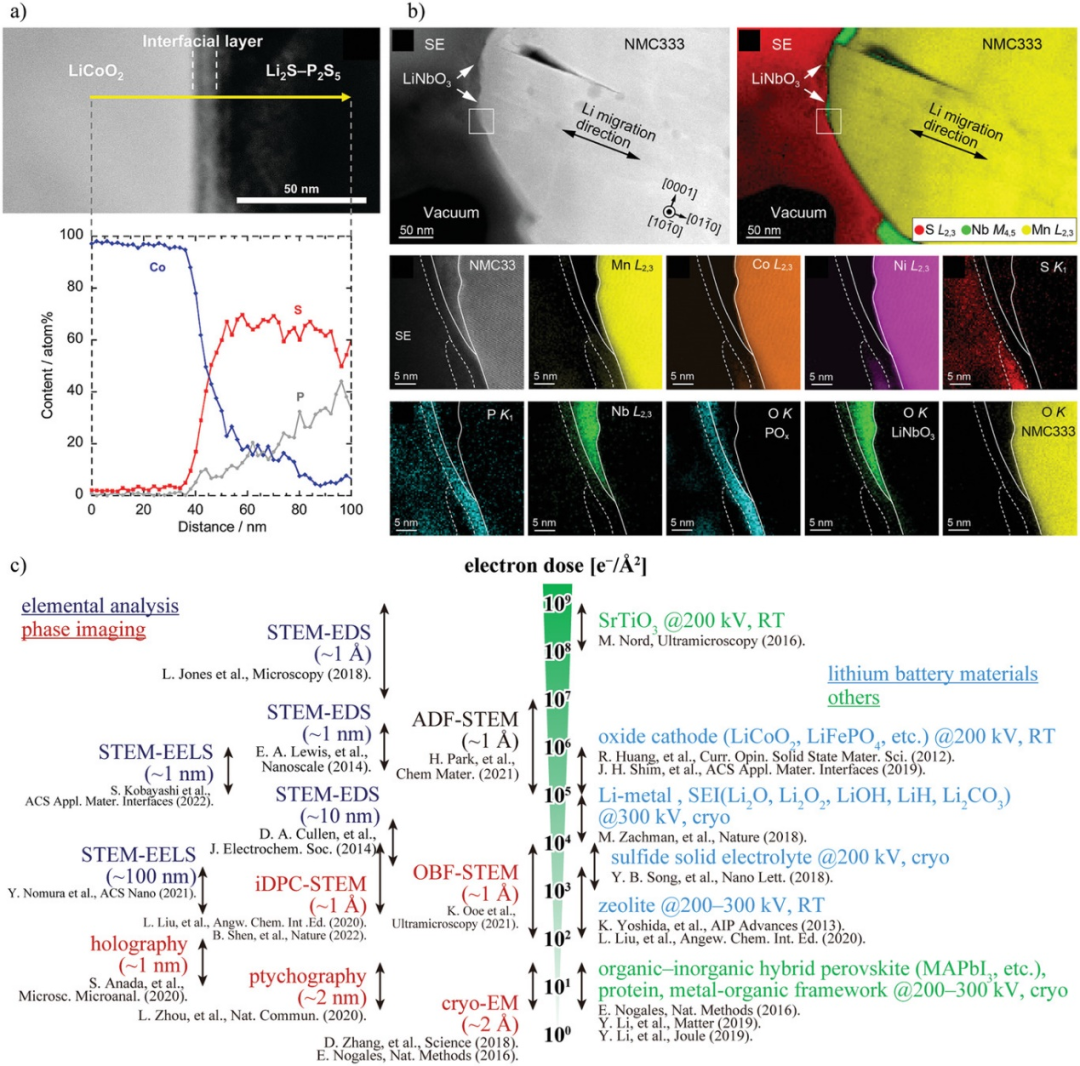
图4. a)LiCoO2/LPS界面初始充电后的高角度环形暗场扫描TEM (HAADF-STEM)截面图和Co、P、S的线扫描。b)通过电子能量损失谱(EELS)测量500个循环后LiNi1/3Mn1/3Ni1/3O2(NCM-333)/LPS界面的HAADF-STEM图像和元素分布。c)电子束敏感材料TEM分析的总电子剂量。@Wiley
由于具有较高空间分辨率,TEM已被应用于正极与硫化物SEs界面的表征。在约10 nm的界面层中,Co、P和S相互扩散,表明Co扩散到LPS中阻碍了Li的传导,增加了界面电阻。在NCM-333与LPS直接接触的区域,Mn、Co、Ni向SE扩散。而在NCM-333被LiNbO3覆盖的区域,过渡金属没有扩散,表明LiNbO3抑制了扩散。此外,在SE表面观察到5 nm的POx层,在NCM-333表面观察到5~10 nm的O耗尽层。
然而,硫化物SEs的TEM分析中存在电子束损伤问题。图4c左侧显示了基于TEM的表征技术使用的单位面积电子剂量。图4c右侧显示了观察典型电子束敏感材料所用的单位面积电子剂量。紫色的元素分析(STEM-EELS和STEM-EDS)手段需要相对较大的电子剂量。对于空间分辨率≈1 Å的STEM-EDS,需要≈108 Å−2的高电子剂量。因此,对于≈10 nm的空间分辨率,可以在≈104 Å−2的剂量下进行STEM-EELS和STEM-EDS。考虑到锂电池材料的电子束敏感性,LCO和LiFePO4等氧化物正极对电子束抵抗能力相对较强,在200 kV的加速电压下,电子束剂量≈105-107 Å−2。而锂金属,二元含锂化合物,如Li2O, LiOH和Li2CO3,以及硫化物SEs对电子束更敏感。它们的结构在电子束剂量为103-105 Å−2时降解。使用空间分辨率为≈1 nm(≈106 Å−2)的STEM-EELS和STEM-EDS进行元素分析将导致SEs分解。
使用电子束相位信息的技术,如积分差分相位衬度(iDPC)STEM、最优明场STEM、叠层成像、电子全息和低温电子显微镜(cryo-EM),在104 Å-2或更低的电子束剂量下,可以获得小于1 nm的空间分辨率。然而,这些技术只能观察原子排列或电位/电场分布,元素分布和电子状态无法被表征。这些技术可以减少EELS分析的电子剂量,使正极/硫化物SE界面具有较高的空间分辨率和较低的损伤。
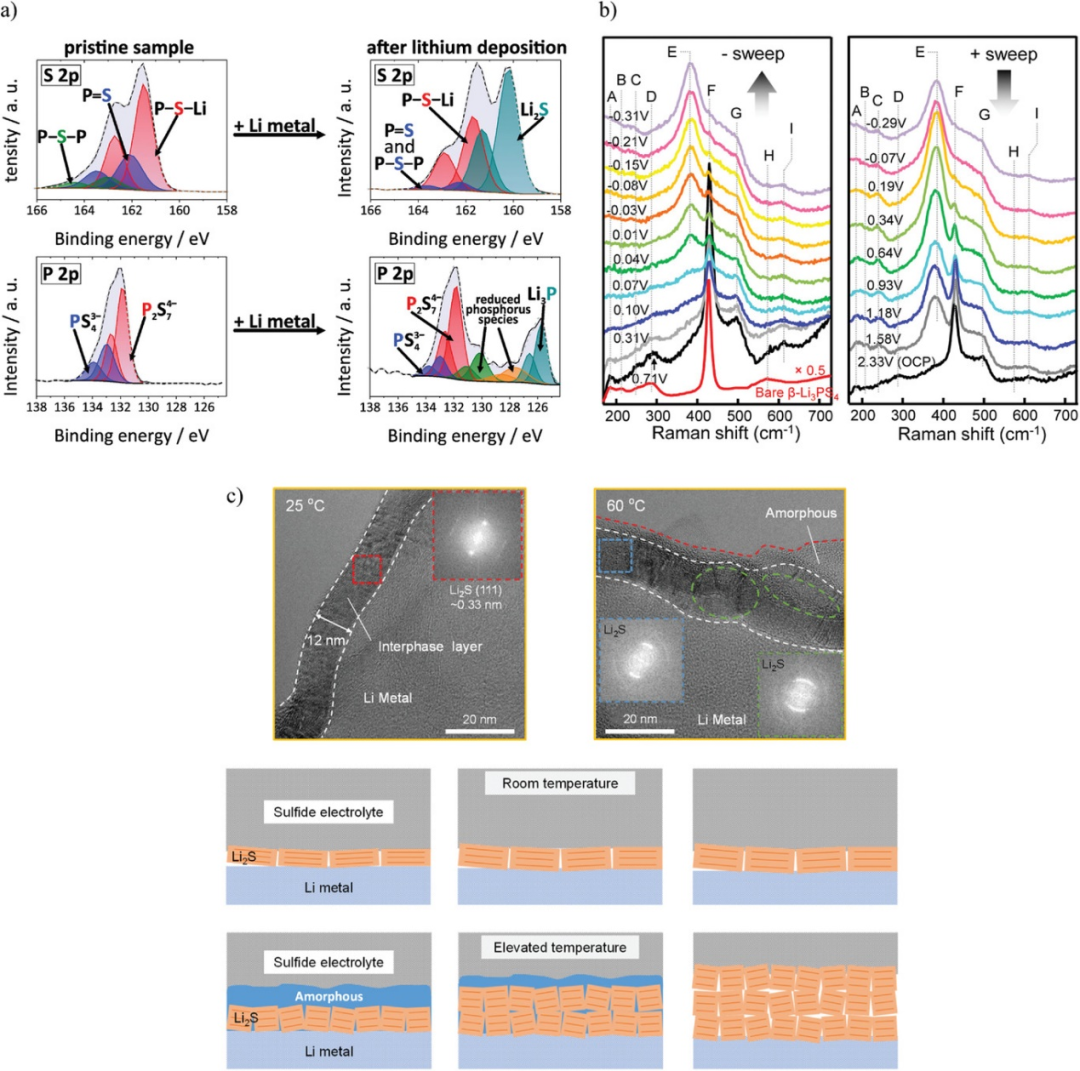
图5. a)Li/Li7P3S11界面S-2p和P-2p光谱。b)锂电镀(左)和剥离(右)过程中β-LPS的原位拉曼光谱。c)Li/Li6PS5Cl界面处SEI的高分辨率低温EM图像。@Wiley
4、SEIs的表征
SEI最常用的表征技术是XPS。图5a为Li7P3S11镀锂前后的S-2p和P-2p XPS谱图。镀锂后,S-2p谱在160 eV处出现一个属于Li2S的峰,P-2p谱在125.5 eV处出现一个属于Li3P的峰。在Li电镀过程中,归因于拉曼光谱β-LPS中PS43−对称拉伸的F峰强度随着电位的变化而降低,而归因于P2S64−中PS3对称拉伸的E峰强度增加。结果表明,在镀锂过程中,β-LPS的还原作用使PS43−转化为P2S64−。这表明PS43−和P2S64−之间的变化是一种与电位有关的氧化还原反应。PS43−和P2S64−之间的差异无法使用EELS轻易区分,因为该技术对β-LPS和P2S64−中存在的P-S键敏感。XPS和拉曼光谱可以用于表征SEI,因为它们可以分别测量微小的电子和结构差异。
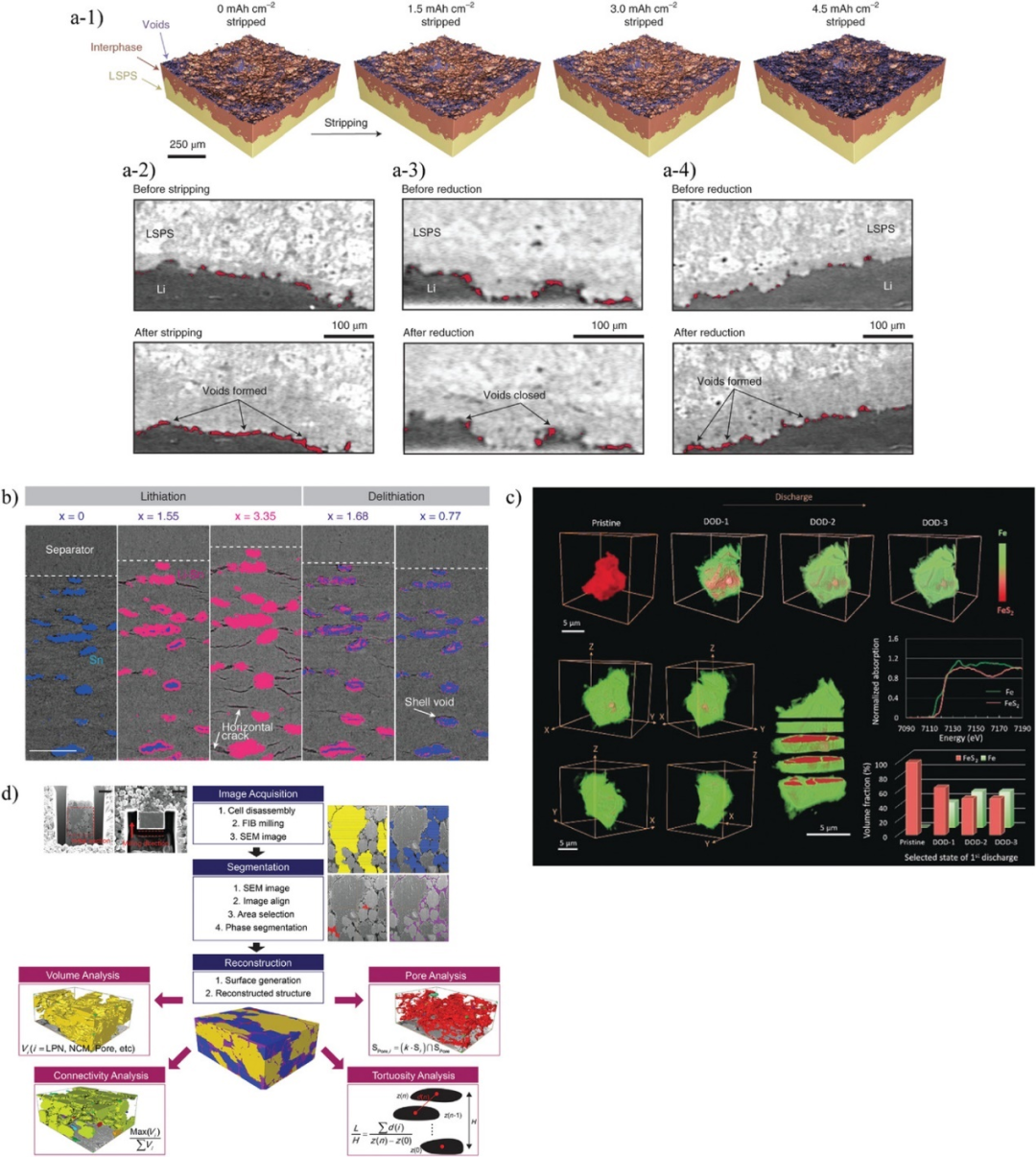
图6. a-1)在1 mA cm−2的剥离过程中,通过原位CT观察Li/LSPS/Li电池中Li/Li10SnP2S12(LSPS)界面上孔洞的演化。a-2)1 mA cm−2下Li/LSPS界面剥离前(上)和剥离后(下)的二维截面图像。a-3)4 mA cm−2还原前(上)和还原后(下)的2D图像。a-4)1 mA cm−2还原前(上)和还原后(下)的2D图像。b)通过原位同步辐射X射线层析显微镜观察Sn/LPS复合电极的结构演化。蓝色和粉色区域分别表示Sn和LixSn。c)FeS2/LPS复合电极中FeS2和Fe的相演化随放电状态的变化。d)使用聚焦离子束和扫描电镜三维重建技术进行定量分析的流程图。@Wiley
5、界面的机械降解
由于活性材料的膨胀和收缩以及SEs分解引起的复合电极机械降解也是一个重要问题。它可能导致:1)离子或电子传导通路弯曲度增加,2)活性材料/SE界面接触面积减小,3)活性材料与SE分离,导致阻抗增加和容量衰减。
动态观察包埋界面上的裂纹和接触损失最常用的技术是X射线计算机断层扫描(CT),它可以在毫米尺度上进行大面积的无损3D观察。图6a通过基于原位同步加速器的CT研究了在外加电压下Li/Li10SnP2S12(LSPS)界面上产生的界面相和孔隙,并定量测量了接触损失,表明界面接触损失和重构是导致电池失效的关键因素。
图6b利用同步辐射CT观察了Sn/LPS复合电极充放电过程中Sn的体积变化和裂纹的形成。充电时,Sn吸收Li,转变为LixSn。Sn在单轴加压方向优先膨胀和收缩。裂纹在水平方向上形成,反映了Sn体积变化的各向异性。这些水平裂纹在析出后闭合,而Sn/LPS界面的界面分层始终存在。因此,CT能够用于原位分析固态锂电池的微观结构和机械降解机制。
与CT相比,基于同步辐射的透射X射线显微(TXM)技术具有更高的空间分辨率,可以用来研究界面处的局部现象。图6c将原位TXM与X射线吸收近缘结构(XANES)相结合,以50 nm的空间分辨率观察FeS2/LPS复合电极中FeS2在充放电过程中的化学状态变化。通过分析Fe-K边XANES光谱,可以发现FeS2向Fe转变,形成核壳结构。在表面形成的Fe层能够作为钝化层阻碍Li的传输。TXM-XANES是一种能够表征活性材料种过渡金属价态变化的技术,其缺点是单次测量所需的时间较长。
图6d使用FIB-SEM可视化了NCM-622/(Li2S)8(P2S5)2(Ni3S2)1(LPN)复合电极的三维结构。通过三维重构,可以量化各组分的体积比、连通性和弯曲度等结构信息以及这些组分之间的界面面积。结果发现,NCM-622/LPN复合电极包含15%的空隙,对电池性能产生了负面影响。另外,导电添加剂的分散性不佳,不能提供有效的电子传导路径。但这种方法存在相位分割问题。SEs、导电添加剂、粘结剂和空隙的扫描电镜强度通常是相似的,因此很难准确分割。可以通过观察扫描电镜图像并手动进行分割,但需要花费大量时间。此问题对FIB-SEM影响较大,但在CT和TXM中也很常见。
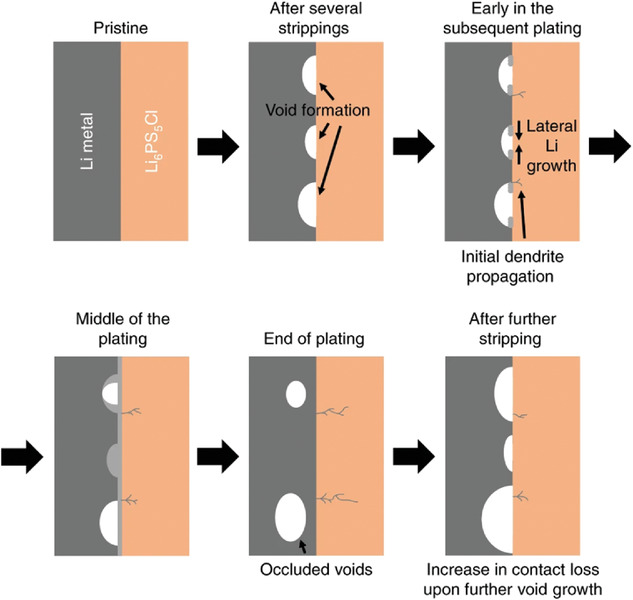
图7. Li/Li6PS5Cl界面在高于临界剥离电流的总电流密度下循环的原理图。@Wiley
6、Li枝晶形成
硫化物SEs内部Li枝晶生长的原因之一是Li与硫化物SEs之间的粘附性较低,导致Li/SE界面电流分布不均匀。因此,Li在电流集中区域附近成核,导致枝晶生长。在镀锂过程中,CCD能够判断导致短路的最大电流密度。然而,在Li剥离过程中也具有CCD。图7为大电流剥离时Li/Li6PS5Cl界面示意图。首先,在界面附近的锂金属层产生孔洞。在随后的循环中,Li沉积在孔隙中。然而,当孔洞非常大时,它们无法被填充,并且闭合的孔洞仍然停留在界面附近。随着循环的进行,这些闭塞的孔隙增加,从而减少了接触面积,增加了非均匀电流分布。这表明,当剥离电流较大时,电池更容易发生短路。

图8. a)在与玻璃LPS断口接触的黄铜尖端电极上电镀锂的原位光学显微镜。b)多晶β-Li3PS4样品断口形貌的SEM图像。@Wiley
光学显微镜是一种动态观察晶体中枝晶和裂纹演变的技术。铜尖电极或SE表面均沉积有Li, SE表面未见枝晶生长。铜尖电极和SE表面均未见Li沉积。相反,在SE中观察到新产生的裂纹轮廓。这表明,Li沿着表面缺陷生长并进入SE。多晶β-Li3PS4切割截面的SEM图像,沿晶界或孔洞的Li枝晶为网状。结果表明,表面缺陷和晶界是影响锂枝晶生长的重要因素。光学显微镜的优点是可以原位地以高时间分辨率观察Li电镀过程。然而,这些仅适用于透明的SE。此外,在纳米分辨率下进行3D分析和观测并不容易。
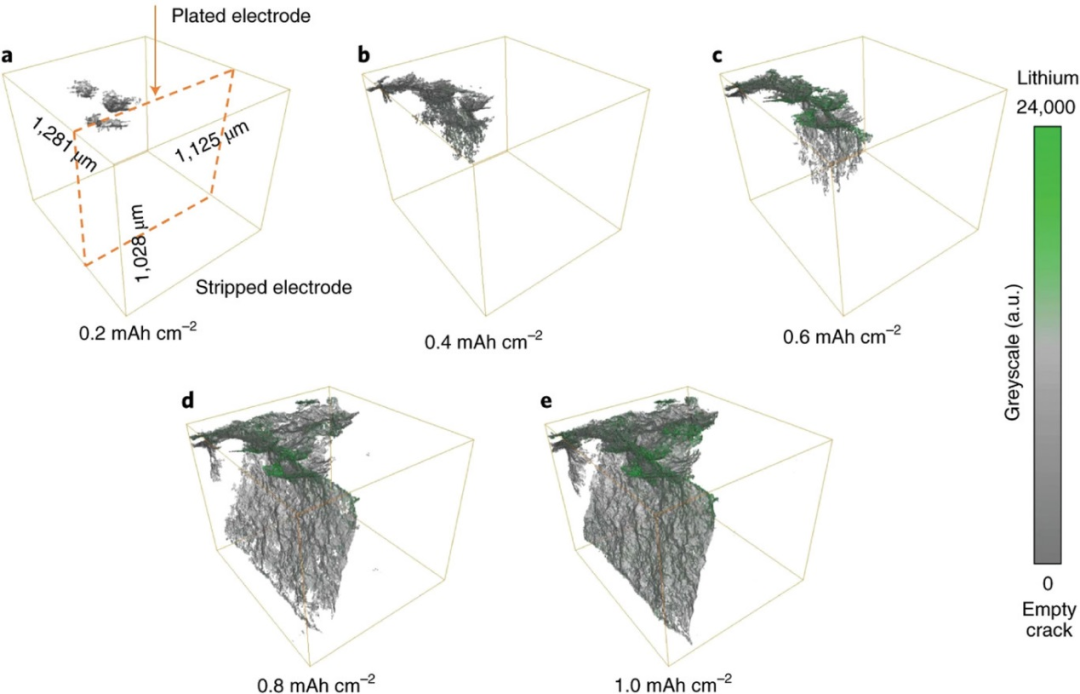
图9. 在a)0.2 mAh cm−2、b)0.4 mAh cm−2、c)0.6 mAh cm−2、d)0.8 mAh cm−2和e)1.0 mAh cm−2电荷通过后,原位CT观察到Li/Li6PS5Cl/Li对称电池中Li枝晶形成。@Wiley
原位CT可以无损地显示在SE层中Li枝晶生长的变化。具有高度相干光束的相位对比成像有效地增强了弱衰减材料的对比度,能够将Li金属与孔洞区分开来。这使得可以独立地观察裂纹扩展和锂电镀。在电池内部,SE裂纹首先在局部电场较大的Li电极边缘形成。横向裂纹沿SE层扩展到另一侧的锂金属表面。图9显示了裂纹和镀Li后裂纹内部沉积Li的原位CT的3D体积渲染图像。当裂纹到达另一侧Li金属表面时,没有发生短路,说明产生裂纹的整个区域没有被锂填充。锂金属通过从后部扩大裂纹,促进裂纹形成,随后锂金属在裂纹内部生长,导致电池短路。
然而,由于CT的空间分辨率较低,很难分析局部结构,如裂纹尖端和枝晶形貌。而具有高空间分辨率以及三维分析能力的FIB-SEM只能观察到一个狭窄的区域。要解决这一问题,可以将CT和FIB-SEM结合使用。首先,原位CT进行无损和3D成像,并确定感兴趣的区域。随后通过FIB-SEM对该区域进行分析,从而能够将电化学性能与纳米尺度下发生的局部现象联系起来。
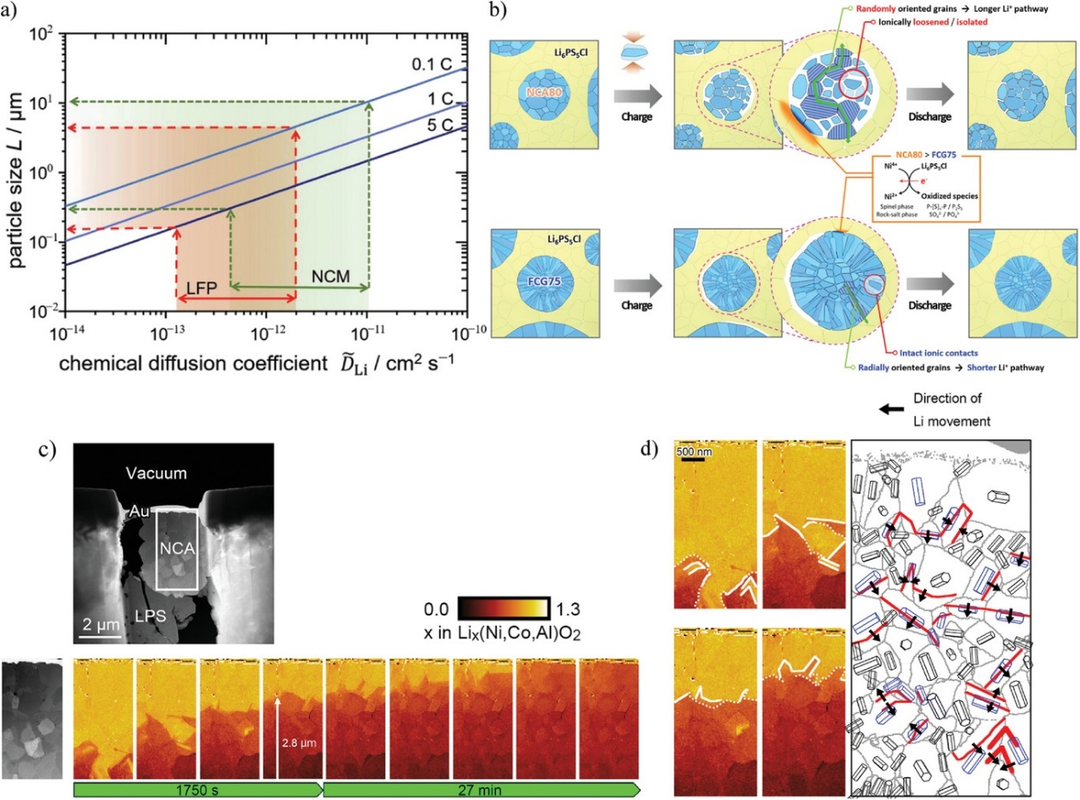
图10. a)根据公式(1),当倍率为0.1,1,和5C时,取决于各自扩散系数范围的最大粒径。b)常规随机取向正极(NCA80)和径向对齐全浓度梯度正极(FCG75)示意图。c)充电过程中NCA次级粒子中Li分布的原位STEM-EELS观测。d)晶粒结构引导下的锂离子运动。@Wiley
7、活性物质中缓慢的Li扩散
Li在活性物质中的扩散缓慢是另一个问题。如果活性物质中的Li扩散跟不上SEs的离子传导,电化学反应就不能有效进行。粒径较大的活性物质由于扩散长度较长,容易使输出特性变差。
图10a显示了每种倍率下的化学扩散系数与粒径之间的关系。从图中可以看出,在1C下,NCM要达到83%的材料利用率,粒径应小于几微米。然而,如果活性物质粒径非常小,则活性物质和SEs不能在复合电极内部高度分散。结果,与SE分离的活性物质比例增加,导致利用率降低。因此,在固态锂电池中,锂扩散对小颗粒尺寸的要求与离子传导路径对大颗粒尺寸的要求相冲突。要实现高功率运行和高活性材料利用率,必须促进活性材料内部的Li扩散。
优化粒子结构有望解决Li扩散问题。层状岩盐正极,如NCM,在晶格中有二维导Li平面(a-b面)。控制初级粒子的取向可以使锂离子在径向上快速扩散。图10b显示了常规随机取向正极(LiNi0.8Co0.16Al0.04O2, NCA80)和全浓度梯度正极(LiNi0.75Co0.10Mn0.15O2, FCG75)的示意图。二次粒子有望在径向上实现快速的Li扩散,此外二次颗粒在径向的体积变化较小,从而抑制了FCG75与SE界面处的机械降解。
对于这样的粒子结构,研究Li扩散系数是很重要的。测定Li化学扩散系数最常用的方法是GITT和EIS。然而,这些方法只能获得平均信息,而不能获得局部信息。使用SIMS、原位Kelvin探针力显微镜(KPFM)和原位STEM-EELS能够测量局部Li扩散。图10c为NCA/LPS/InLi电池充电过程中NCA二次粒子内部Li浓度变化的原位STEM-EELS图。Li从NCA中脱出,实现了对Li扩散的分析。图10d显示了Li扩散方向与晶体取向之间的关系。黑色箭头所示的Li扩散方向在三方晶系NCA的基面(a-b平面),说明Li沿二维导电平面扩散。因此,原位STEM-EELS能够以纳米级的空间分辨率分析活性材料内部的局部Li扩散。
05
成果启示
1)硫化物SEs与正负极都存在热力学不稳定性问题。XPS、ToF-SIMS和拉曼光谱都有望揭示界面处的分解产物。电子显微镜能够以更高的空间分辨率分析分解产物的晶体结构、确切组成和空间分布,但其存在电子辐照损伤。低剂量电子显微镜技术,如低温电镜,结合原位分析手段对于理解分解机制十分重要。
2)由活性物质体积变化和硫化物SEs分解引起的机械降解会破坏离子和电子传导通路,导致阻抗增加和容量衰减。原位CT分析能够动态观察加压条件下复合电极的三维结构,从而研究微结构与电池性能之间的关系。然而,CT的空间分辨率不足。原位CT或TXM结合具有更高空间分辨率的FIB-SEM有望提供更详细的微观信息。
3)锂枝晶生长也是一个问题。利用原位表征技术,能够可视化裂纹与枝晶扩展路径。然而,这些因素之间的相关性还不完全清楚。使用具有高空间分辨率的原位三维分析方法将阐明这些问题。
4)锂在活性材料中的缓慢扩散可能是限制硫化物基固态锂电池功率密度的主要问题。通过设计特殊的晶粒结构有望促进锂离子扩散,如全浓度梯度正极。原位KPFM和原位STEM-EELS可以在纳米尺度上观察活性材料内部的局部Li扩散。
审核编辑 :李倩
-
全固态锂电池领域的研究及实际应用2022-09-27 4525
-
锂电池VS聚合物锂电池,谁才是未来的主角?2018-08-17 7330
-
日本电池展上固态锂电池大放光彩 将会成为动力电池的下一个风口2019-03-04 1294
-
全固态电池的三大技术路线:氧化物/硫化物/聚合物2020-12-25 5851
-
氧化物硫化物聚合物全固态电池的三大技术路线2021-03-18 2270
-
硫化物固态电解质与氧化物正极的热稳定性2022-11-08 6512
-
固态电池电解质的分类及性能对比2022-11-30 20850
-
利用三甲基硅化合物改善硫酸盐固态电解质与阴极材料的界面稳定性2023-11-01 3419
-
固态电池三大技术路线的优缺点2023-11-21 8912
-
铜集流体是否适用于硫化物全固态电池?2024-01-10 3332
-
三菱综合材料成功开发一种全固态锂电池材料的制造新技术2024-02-27 2245
-
孙华军:硫化物固态电池预计2027年起步入示范应用阶段2024-09-02 2531
-
华为公布硫化物固态电池新专利,固态电池技术加速发展2024-11-07 3099
-
清华深研院刘思捷/港科大Kristiaan Neyts最新AEM封面文章:硫化物复合固态电解质2025-01-07 1504
-
清华大学:自由空间对硫化物固态电解质表面及内部裂纹处锂沉积行为的影响2025-02-14 1236
全部0条评论

快来发表一下你的评论吧 !
