

首次MoS₂层间原位构建静电排斥实现超快锂离子传输
描述
高理论容量和独特的层状结构使MoS₂成为一种很有前途的锂离子电池负极材料。然而MoS₂层状结构的各向异性离子输运和其较差的本征导电性,导致差的离子传输能力。针对这些问题,本文提出由Co²⁺取代Mo⁴⁺在MoS₂层间原位构建层间静电排斥,其可打破层间范德华力的限制,制备单层MoS₂,从而建立各向同性的离子传输路径。同时,掺杂的Co原子改变了单层MoS₂的电子结构,从而提高其电导率。重要的是,掺杂的Co原子可以转化为Co纳米颗粒,从而产生空间电荷区以加速离子传输。因此,Co掺杂的单层MoS₂展示出了超快的锂离子传输特性。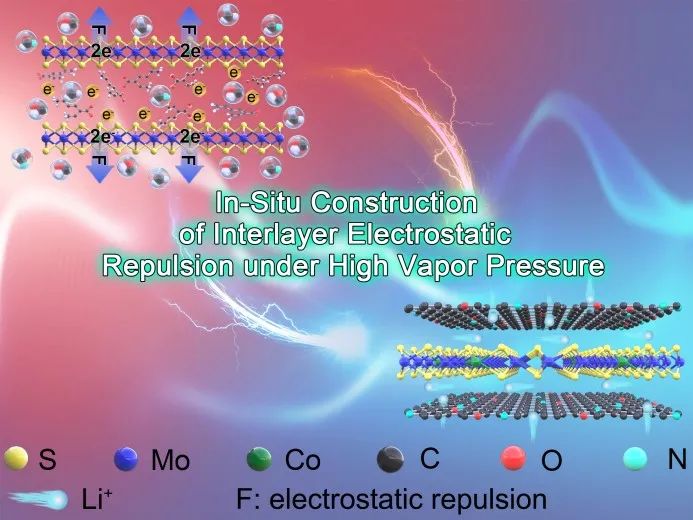
本文亮点
1. 在高压气相下,首次利用有机离子液体在MoS₂层间原位构建了静电排斥,成功制备了Co掺杂单层MoS₂。
2. 掺杂的Co原子从根本上降低了单层MoS₂的带隙和锂离子扩散能垒,并且可以转化为超小的Co纳米颗粒(~2nm),在转化反应过程中产生强烈的表面电容效应。
3. 作为锂离子电池负极材料,Co掺杂单层MoS₂表现出超快的离子传输能力以及超高容量和出色的循环稳定性。
内容简介
高理论容量和独特的层状结构使MoS₂成为一种很有前途的锂离子电池负极材料。然而MoS₂层状结构的各向异性离子输运和其较差的本征导电性,导致不可接受的离子传输能力。针对这些问题,南方科技大学赵天寿院士和韩美胜副研究员等人提出由Co²⁺取代Mo⁴⁺在MoS₂层间原位构建层间静电排斥,通过调节Co²⁺掺杂量可以调控层间静电斥力大小,当其远大于层间范德华力时,可以获得单层MoS₂,从而建立各向同性的离子传输路径。同时,DFT模拟揭示了掺杂的Co原子改变了单层MoS₂的电子结构,从而提高其本征电导率,降低了锂离子扩散能垒,加速了锂离子传输速度。
重要的是,掺杂的Co原子可以转化为超细小的Co纳米颗粒,从而产生强烈的表面电容现象,从而加速锂离子传输。在半电池中,在0.1 A/g的电流密度下,其具有1661.6mAh/g的容量,在20A/g时容量仍高达1063.3mAh/g。在全电池中,在4C的电流密度下,其在11.5min的充电时间,能量密度可以高达136.2 Wh/kg,维持了76.6%的能量保持率,证明了其具有超快的锂离子传输特性。
图文导读
I Co掺杂单层MoS₂形成机制
本文使用四硫代钼酸铵、DMF和金属离子液体异辛酸钴作为前驱体,将其封装在密闭装置中加热,使前驱体热解,金属离子液体中的Co²⁺会原位取代MoS₂中Mo⁴⁺的位置,使形成的MoS₂带有负电荷,根据同种电荷相互排斥理论,在MoS₂层间会产生静电斥力,在静电斥力和前驱体热解产生的气态基团的共同作用下,单层MoS₂形成(图1a-d)。为了说明这一过程,进行了DFT模拟计算。从图1e-g可以看出,随着Co²⁺掺杂量的增加,MoS₂层间距会逐渐变大,当Co和Mo原子比为1:2时,层间距扩大到2.36nm,此时层间范德华力消失,单层MoS₂形成。从图1h-k可以看出,Co掺杂的单层MoS₂均匀分散到了N,O共掺杂的碳基底中。
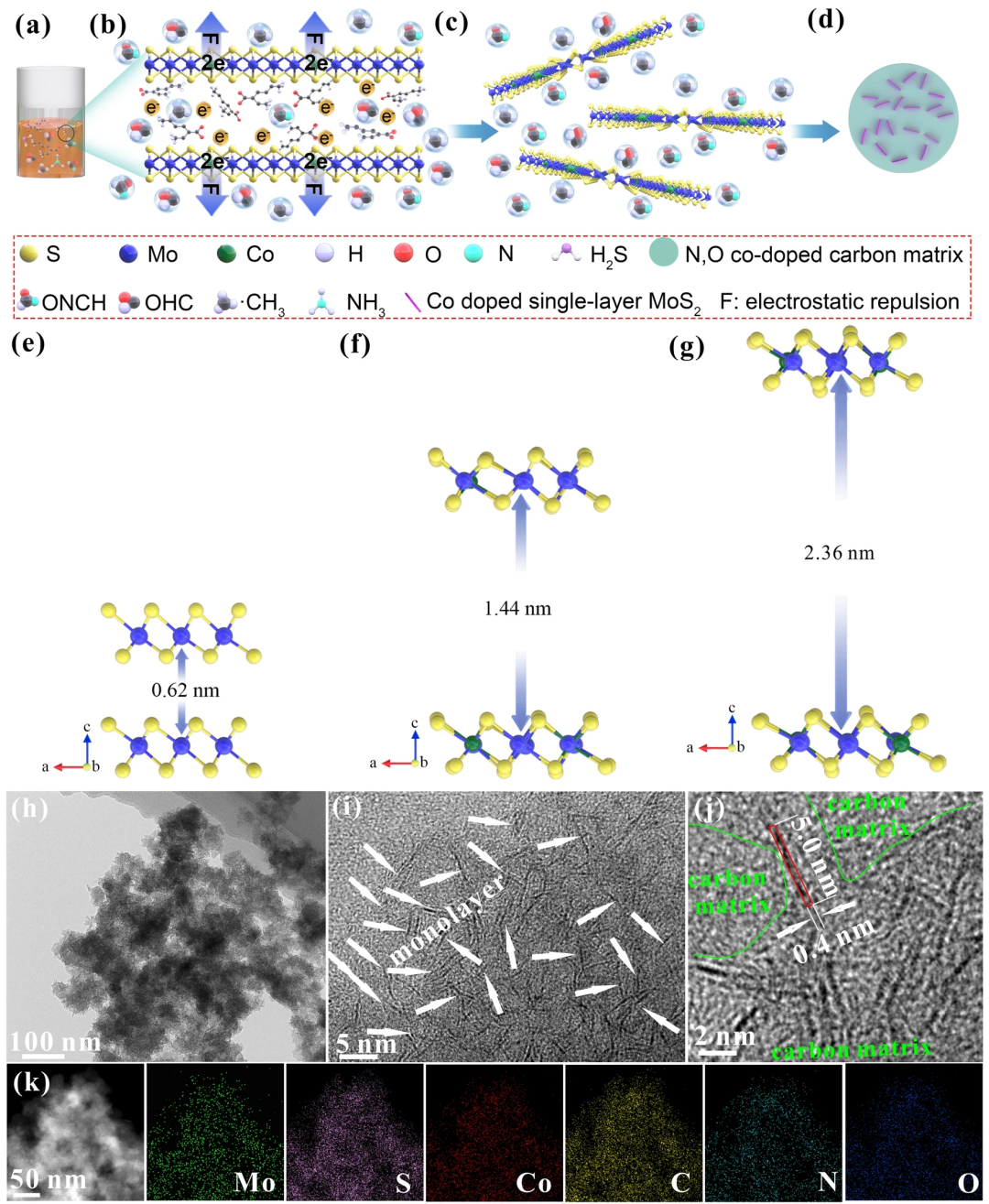
图1. a-d单层MoS₂的形成机制,e-g DFT模拟计算结果,h-k TEM表征。
II Co掺杂单层MoS₂材料结构和成分分析
CoMoS₂/C-I为低Co掺杂量获得的样品,CoMoS₂/C-II为适中Co掺杂量获得的样品,CoMoS₂/C-III为高Co掺杂量获得的样品。从图2a可以看出,随着Co掺杂量增加,(002)逐渐消失,证实了单层MoS₂的形成,同时可以看到过量的Co掺杂除了单层MoS₂的形成,还获得了杂质相Co₃S₄,说明了适当的Co掺杂对获得纯单层MoS₂的重要性。图2b-h的Raman和XPS结果进一步证明了以上结果。图2i的TGA图可以看出,随着Co掺杂量的增加,碳含量逐渐增加,说明异辛酸钴中的碳链在高压气相的作用下更容易转化为碳材料,从而增加碳含量。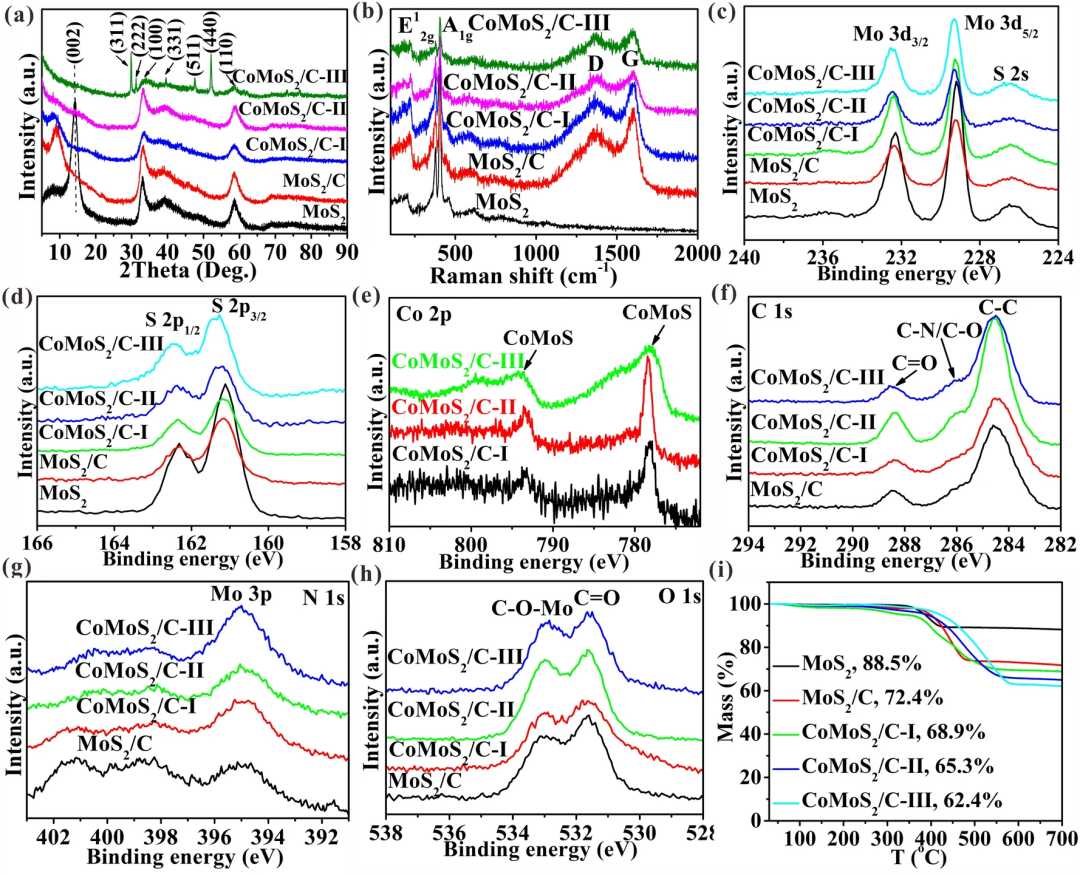
图2. a XRD, b Raman, XPS 图谱:c Mo 3d, d S 2p, e Co 2p, f C 1s, g N 1s, h O 1s, 和i TGA。
III Co掺杂单层MoS₂电化学性能表征
从图3a即首次充放电曲线可以看出,合成的CoMoS₂/C-II(单层MoS₂)样品充放电容量最高达到1512.9 mAh g⁻¹ 。图3b循环曲线显示CoMoS₂/C-II在100次循环后仍然保持了较高的容量1504.3 mAh g⁻¹,几乎与首次充放电容量持平,其性能与MoS₂/C and CoMoS₂/C-I类似。说明CoMoS₂/C-II经多次循环后仍能保持其电极完整性且体积膨胀较小。纯的MoS₂循环性能最差,主要是由于缺乏单层结构和N,O掺杂的碳基体。从图3d看到CoMoS₂/C-II其电荷转移电阻最小,使其比其他样品具有较高的Li⁺传输速率,使其在20 A g⁻¹的电流密度下,容量高达1063.6 mAh/g(图3c)。此外,图3f-h分别为CoMoS₂/C-II样品在0.1A/g,1A/g和5A/g的循环稳定性曲线,可以看出,其具有非常好的循环稳定性。CoMoS₂/C-II无论在容量、倍率性能和稳定性方面都高于之前报道的MoS₂基负极材料(图3i)。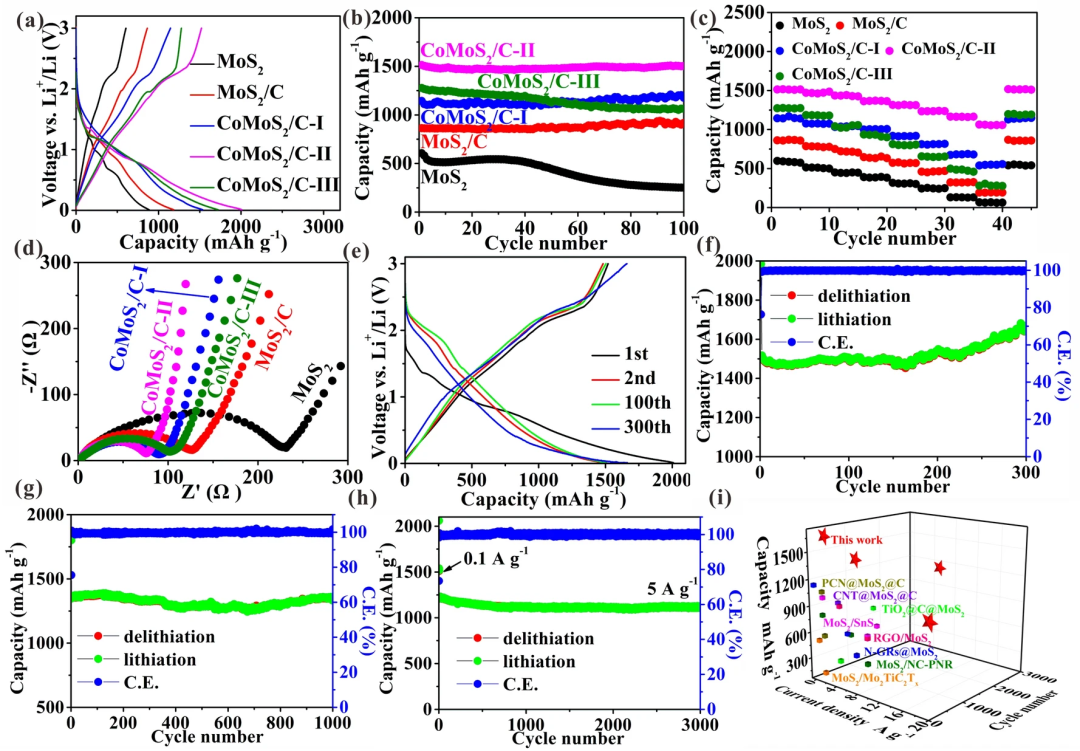
图3. a 充放电曲线,b 循环曲线,c倍率曲线,d EIS曲线,e 单层MoS₂充放电曲线, f 单层MoS₂ 0.1 A/g循环曲线, g 单层MoS₂ 1 A/g循环曲线,h 单层MoS₂ 5 A/g循环曲线,i 性能对比。
IV Co掺杂单层MoS₂转化反应机制
图4a,b可以看到放电到0.01V时Co掺杂MoS₂的层状结构彻底消失,转化为了大量的晶体纳米颗粒,颗粒尺寸约为2nm,图4c的衍射结果可以看到,纳米颗粒为Co,Mo和Li₂S。图4d可以看到在放电到0.01V时,Mo3d的峰向低结合能的方向移动,228.3 eV代表了Mo纳米颗粒的形成,在充电到3V时,Mo3d峰进一步向低结合能方向移动,说明了在其表面空间电荷区的形成。图4e,可以看到在放电到0.01V时,Co2p的峰向低结合能的方向移动,777.8eV代表了Co纳米颗粒的形成,在充电到3V时,Co2p峰进一步向低结合能方向移动,说明了在其表面空间电荷区的形成。为了说明空间电荷区的形成,电极在不同充电状态的磁滞回线是被测试的(图4f)。从图4f可以看出,原始电极的磁化强度基本为0,而在首次放电到0.01V后,电极磁化强度迅速增加,主要是由于磁性Mo和Co纳米颗粒的形成,在充电到3V后,电极磁化强度进一步增加,说明Mo和Co表面形成了空间电荷区。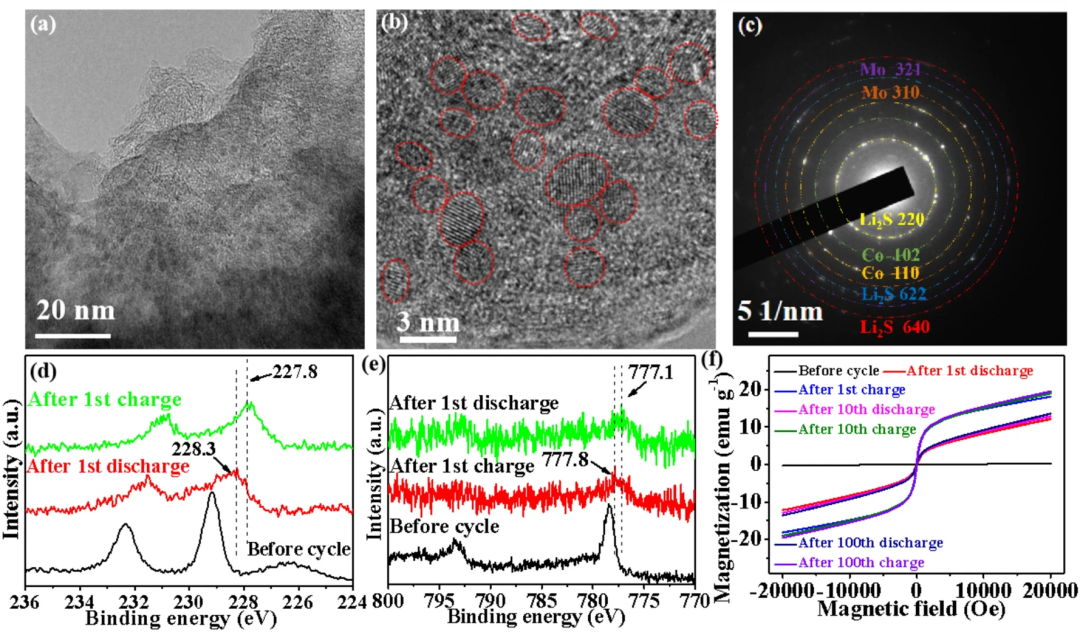
图4. a,b Co掺杂单层MoS₂电极放电到0.01V时TEM照片,c SAED,d Mo 3d的XPS谱,e Co 2p的XPS谱, f 电极在不同循环圈数电极的磁滞回线。
V Co掺杂单层MoS₂全电池性能
本文选择磷酸铁锂做正极,Co掺杂单层MoS₂材料为负极,组装全电池,在组装之前,Co掺杂单层MoS₂材料在半电池中先预循环三圈,以提高全电池首次库伦效率。从图5a,b可以看出,在0.1 C循环100圈后,容量高达164.4 mAh/g,容量保持率为95.1%。在1 C循环200圈后,容量仍高达133.9mAh/g,容量保持率90.2%(图5c)。此外,其表现了突出的快速充电能力,在4C时容量可达131.8mAh/g,保持0.1C所获得容量的80.2%(图5d,e),对应的能量密度为136.2Wh/kg,保持0.1C能量密度的76.6%(图5f)。在4C电流密度下,循环500圈,容量保持率高达80.2%,足以说明其具有快速充电能力的同时,还具备突出的稳定性(图5g)。组装的一个扣式电池可以点亮59个LED灯,且点亮时间可持续2小时,说明了其具有良好的应用前景。
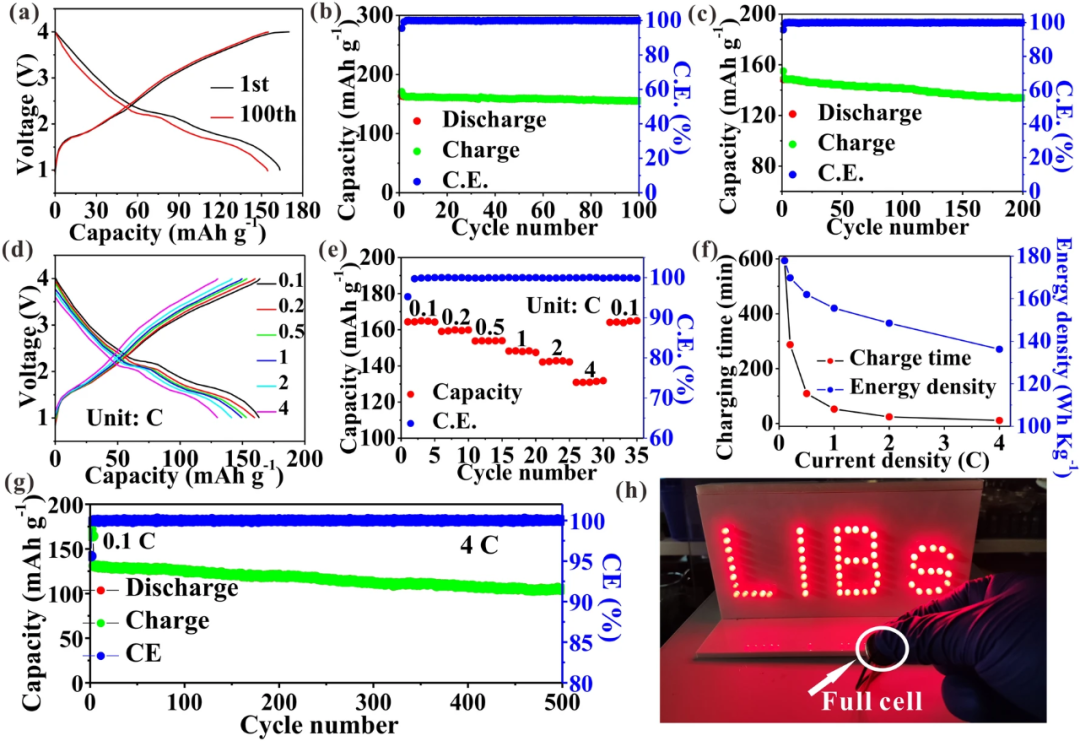
图5:a 0.1 C充放电曲线,b 0.1 C循环曲线, c 1 C循环曲线, d,e 倍率性能, f充电时间和能量密度关系,g 4C循环性能,h LED灯驱动演示。
VI DFT模拟计算
为了说明Co掺杂对单层MoS₂锂离子扩散能垒和电子结构的影响,进行了DFT模拟。从图6a-c可以看到,Co掺杂后极大降低了单层MoS₂的锂离子扩散能垒,从图6d可以看出,Co掺杂后将单层MoS₂的带隙从1.3eV降低到了0eV,即Co掺杂后将单层MoS₂的半导体特性改为了金属性,可以极大促进电子的转移。图6e可以看到单层MoS₂具有各向同性的锂离子存储行为,而多层MoS₂具有各向异性的锂离子存储行为,锂离子需绕道到层间进行存储(图6f),这必将增加锂离子扩散能垒,正如图6g计算的那样,多层层间传输锂离子扩散能垒要高于单层。即本文获得的Co掺杂单层MoS₂与碳的复合材料具有开放性的锂离子存储行为(图6h),且转化反应形成了大量的Mo和Co纳米颗粒,在其表面可以建立强烈的空间电荷区,从而加速锂离子传输速率和增加锂离子存储数量(图6i),从而极大地增加倍率性能和容量。
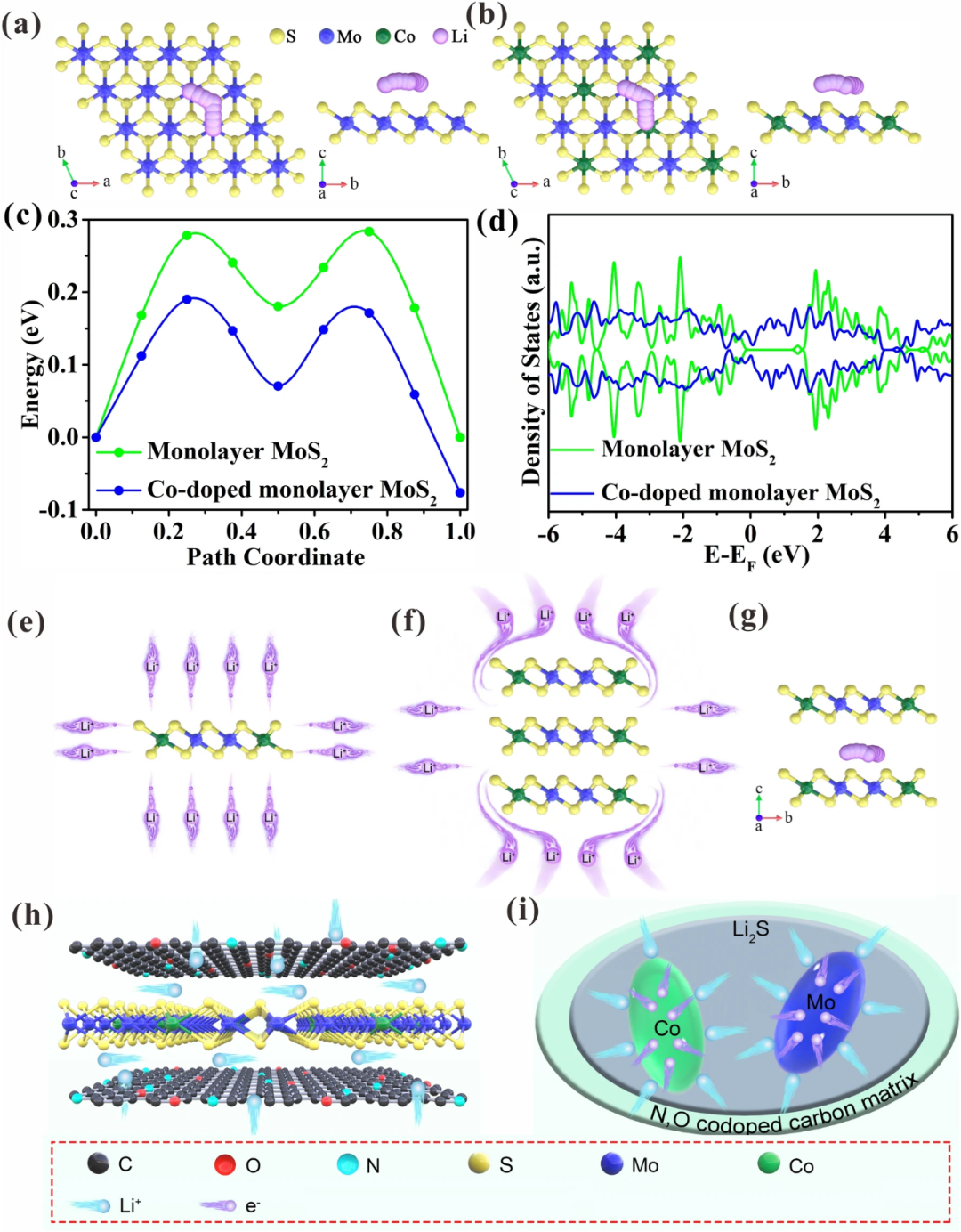
图6. a 纯单层MoS₂锂离子扩散模型,b Co掺杂单层MoS₂锂离子扩散模型,c 扩散能垒,d DOS结果,e Co掺杂单层MoS₂锂离子扩散示意图,f Co掺杂多层MoS₂锂离子扩散示意图,g 多层MoS₂锂离子扩散模型,h 插入反应过程中锂离子扩散路径,i 转化反应锂离子存储示意图。
审核编辑:刘清
-
聚合物锂离子电池的构成2013-06-06 3471
-
锂离子电池的基本组成及关键材料2013-07-03 4401
-
锂离子电池的工作原理和使用注意事项2014-10-29 6493
-
如何检测锂离子电池保护板中MOS管的好坏?2015-06-07 6278
-
锂离子电池和锂电池的区别2015-12-28 5861
-
锂离子电池的制造概述2017-02-27 4702
-
锂离子电池充放电设备的保护2018-09-27 3374
-
锂离子动力电池隔膜浅谈2018-10-10 5353
-
锂离子电池SEI膜的性能影响2019-05-24 3154
-
锂离子电池简介2020-11-03 2904
-
石墨烯基锂离子电池产品有哪些2017-10-23 3288
-
静电是如何击穿MOS管的2021-07-21 4362
-
防止过充的有害影响的关键因素是均匀的锂离子脱出2022-09-16 2155
-
万立骏&郭玉国:聚硫化物排斥的原位固态化界面2023-01-16 1952
全部0条评论

快来发表一下你的评论吧 !
