

硼酸盐基可充钙硒电池研究
描述
研究背景
钙金属的氧化还原电位低,钙化合物丰度高,因而钙离子电池(CIB)是一种很有前途的下一代储能系统。为了继续开发CIB,需要发现高能量密度的钙离子正极来实现实际的能量密度值。然而,由于缺乏能够进行可逆钙镀层和剥离的功能性电极材料和合适的电解质,CIB的发展受到了阻碍。最近的报告强调了电解质的重要性,这些报告强调了可逆钙镀层和汽提方面的进展。
最近,新合成的硼基钙盐(Ca(B(hfip4)2在二甲醚电解液中有所报道,表现出的高离子电导率(∼8 mS cm–1),高氧化稳定性(4.5 V vs. Ca2+/Ca),高效的钙剥离和沉积,以及与Ca金属负极和其他正极材料的良好化学相容性。总之,这些属性满足了钙金属电池电解质所需的大部分先决条件,在钙金属上形成保护膜,支持长时间循环。尽管插层材料的结构完整性有利于较长的循环寿命,但观察到的比容量仅限于每个过渡金属阳离子最大值0.5 个钙离子。另一方面,转换电极可以通过利用金属的所有可能的氧化还原状态作为高能量密度CIB的组分来提供高比容量。
成果简介
近日,美国阿贡实验室的John T. Vaughey等人报告了使用Se 作为CIB的高容量正极材料,在Ca金属电池中该材料在室温下通过的转换机制运行。Se电极在 100 mA g–1 时的可逆比容量为 180 mA h g–1,放电平台接近 2.0 V(vs. Ca2+/Ca),测试时使用基于盐的电解质四(六氟异丙氧基)硼酸钙 (Ca(B(hfip4)2),使用了 1,2-二甲氧基乙烷 (DME)溶剂和钙金属负极。并且,作者利用基于同步辐射光源的测试技术研究了钙和Se之间的可逆电化学反应,并讨论了可能的反应机理。
研究亮点
使用Se作为非水钙离子电池正极材料,配合Ca(B(hfip4)2/DME电解液实现可逆运行。
电池在 100 mA g–1 时的可逆比容量为 180 mA h g–1,放电平台接近 2.0 V(vs. Ca2+/Ca)
使用同步辐射原位X射线衍射(XRD)、X射线吸收光谱(XAS)和能量色散X射线光谱(EDS)研究了Se电极的转换过程。
图文导读:
使用Ca(B(hfip4)2在DME中的溶液作为电解液,并使用循环伏安法(CV)进行测试,以验证其作为钙金属电池电解质的稳定性(图1)。CV数据证实,在Au(图1a)和Ca(图1b)工作电极上的可逆钙镀层和剥离都存在低过电位,并支持其在全电池配置中的使用。
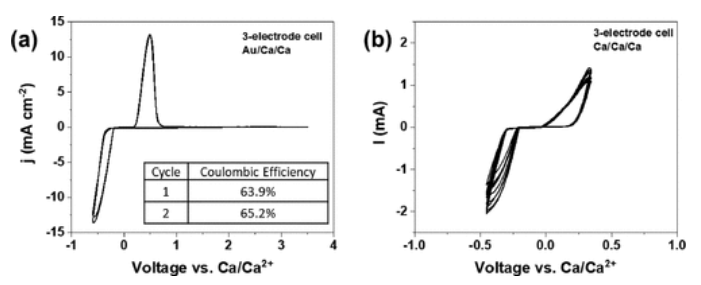
图1. 电池在以(a)金(两个循环)和(b)Ca工作电极(10个循环)。钙棒用作对电极和参比电极。循环伏安法在50 mV s–1下运行扫描速率。
Se/AC 正极和钙金属负极电池(图2 a,使用了世伟洛克型电池)的放电-充电曲线如图 2 b 所示。Se/AC 电极的首次循环放电容量为 235 mA h g–1充电容量为 180 mA h g–1电流密度为 100 mA g–1 时,分别对应于插入 0.35 和脱出 0.27 mol 的 Ca2+/Se。电极在2.0和2.6 V附近显示出明确的放电和电荷平台(与钙相比)2+/Ca),与之前的报告相比,缺乏明显的电压平台,表明负极对总容量的电容贡献很大。基于假设端元CaSe的形成能(−2.26 eV/原子),反应Ca + Se CaSe的平均电压可以计算为2.26 V(vs. Ca2+/Ca),接近放电和电荷平台的中点(∼2.3 V (vs. Ca2+/Ca))。
此外,放电和充电过程中的单电压平台表明氧化还原反应是单步反应,而不是多个逐步反应。这与以前的Se电化学研究不同,后者观察到多个电压平台。氧化还原反应在dQ / d V曲线中得到证实,氧化还原峰之间的差异为0.68V(图2b)。虽然电压迟滞高于Li-Se系统中观察到的电压迟滞(0.1-0.2 V),但它与其他多价离子电池系统(Zn-Se和Mg-Se)相当,这可能表明电荷转移或多价离子扩散相关现象对该机制很重要。在循环时,Se/AC电极在相同的电流密度下评估几个周期时,显示出显着的容量衰减(图2 c)。与报道的硫基体系相比,这归因于正极问题(例如,溶解和粉碎)、负极表面钝化或可能的组合。容量衰减问题将在此背景下讨论,并将在以下研究中进一步研究。
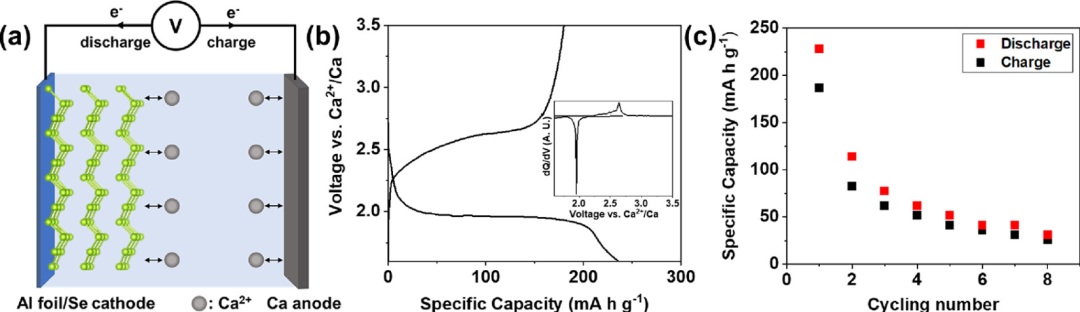
图2. (a) Se/AC正极和Ca负极的钙金属电池示意图。(b) 100 mA g–1 时Se正极的恒电流放电-电荷曲线在室温下和相应差分容量图。(c) Se/交流正极在100 mA g–1时的循环性能在室温下。
文中更详细地研究了Ca-Se反应机理,利用原位XRD研究了Se电极在循环过程中的相演变。用于这些操作研究的冲孔纽扣电池设计如图3 a所示。图3 b显示了操作XRD测量期间原位纽扣电池的电压曲线。请注意,用于此特定数据收集的原位纽扣电池的观测电荷容量略低于通常使用的世伟洛克电池。操作电池显示出略高的电压迟滞,可能是由于电池设计包含更多组件,这可以增加接触电阻,从而增加电压电位。图3 c,d中显示的完整XRD图谱是在原位测量期间观察到任何峰变化的放大XRD图谱。当Se与Ca2+反应时,观察到Se峰强度降低。当电极放电深度达50%时,Se XRD峰大部分消失,在进一步放电或充电时没有观察到其他变化。基于这些观察结果,我们假设在这些转化反应下形成的放电和电荷产物是无定形或纳米晶。
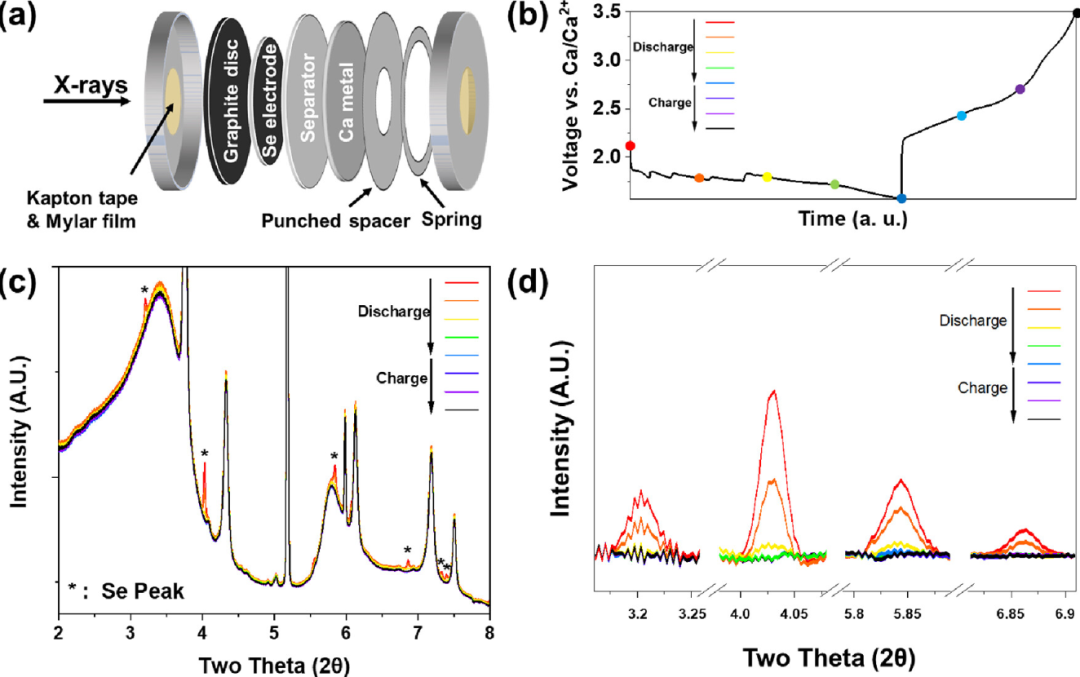
图3. (a) 用于测试的纽扣电池示意图。(b) 原位SXRD测量期间的电压曲线。(c) 在放电和充电期间测量的全原位同步加速器XRD图谱。(d) 在放电和充电过程中原位放大SXRD模式。对XRD图谱进行了背景扣除。
为了深入了解Se/AC电极的电荷存储机理,通过原位同步辐射XAS测量研究了第一个循环的Se氧化还原反应和局部结构变化。原位XAS可能非常有意义,特别是对于XRD信息有限的非晶相。图4 a是XAS测量期间原位电池的电压曲线。图4 b和图4 c分别存在于放电和充电过程中的原位Se K边缘X射线吸收近边缘结构(XANES)谱图,相应的一阶导数图如图4 d(放电)和图4 e(充电)所示。Se K边缘吸收是由于电子从1s态跃迁到未占据的4p态而发生的。在放电和充电过程中观察到XANES光谱的可逆变化。
由于价4p轨道的电子群增加,白线峰的强度在放电期间降低,反之亦然。图4 b,c中与Se还原和氧化相关的边缘偏移看起来很小。边缘的能量位置可以确定为XANES光谱一阶导数的最低能量峰值。在图4d,c中,确认由于Se的减少,Se K边缘向较低能量移动,并且在充电过程中又向高能量移动。然而,完全充电状态与其原始状态并不相同,表明只有部分可逆性,并且与前面提到的第一个循环电化学测试一致。我们怀疑这与充电容量相对于放电容量的下降有关(图4a),并且可能还与原位测试装置有关。
特别令人感兴趣的是,XANES光谱显示了等吸点,表明Se在循环过程中经历了两相反应。为了检查反应机理,测量了假设的最终产物CaSe的Se K边缘XANES光谱(图4b)。CaSe的Se K边缘XANES光谱与为实验样品收集的原位XANES光谱有很大不同,如图4 b所示。这表明在我们的实验条件下,完全钙化的材料CaSe可能不存在,但它很可能是组成CaSex (x > 1),这与测量的容量一致。为了更多地了解放电正极相的性质,在循环过程中使用原位扩展X射线吸收精细结构(EXAFS)光谱研究了Se周围的局部环境。图4 f和图4 g分别是放电和充电过程中的原位Se K边缘EXAFS光谱。在傅里叶变换EXAFS中,2至2.5 Å(相位未校正)的峰是Se-Se第一邻键,2.5至3 Å的峰则是Se-Se第二相邻键。在循环过程中,在EXAFS光谱中观察到局部结构的部分可逆性,这与XANES结果一致。
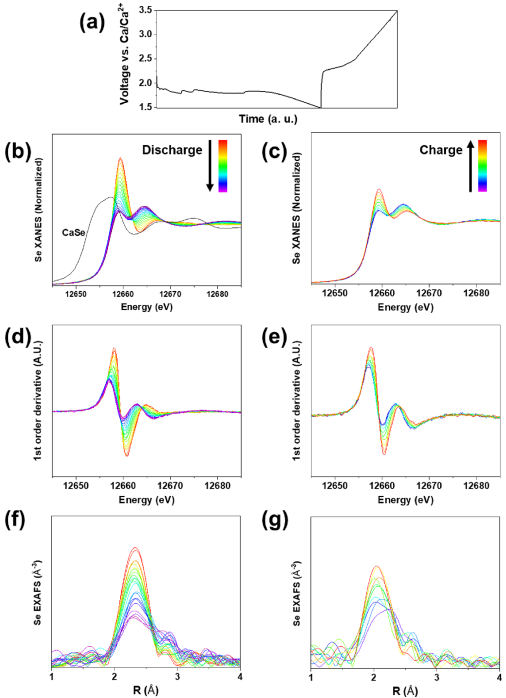
图4. (a) 原位XAS测试期间的电压曲线。(b)放电和(c)充电期间的原位Se K边缘XANES光谱。CaSe的Se K边缘XANES光谱在(b)中。(d) 放电和 (e) 充电期间原位 Se K 边缘 XANES 光谱的相应一阶导数图。(f) 放电和 (g) 充电期间的原位Se K 边缘 EXAFS 光谱。
对电极进行EDS分析,以确认循环过程中Ca2+插入和脱出的程度(图5)。用乙腈洗涤Se/AC电极,以确保没有钙电解质盐的残留物残留在表面上,即使在洗涤过程中存在活性物质损失的可能性。EDS光谱以Se信号归一化。放电的Se电极显示Ca Kα的Ca信号为3.7 keV,Ca Kβ的Ca信号为4.0 keV。充电后,Ca信号相对于Se峰下降,支持相对于放电样品脱去Ca。电极中Ca的含量基于EDS定量结果并根据EDS样品的电化学计算得出,如图5所示,变化趋势大致一致。
总结与展望
总之,Se已被评估为钙离子电池中的转化正极,并在非水钙基电解质中使用钙金属负极进行研究。本研究发现Se电极准可逆地转化为CaSex(x ≈ 0.3),在 100 mA g–1初始放电容量为235 mA h g–1 时,放电平台约为 2.0 V(vs. Ca2+/Ca)。与典型的多价电池电极相比,Se正极显示出明确的电压平台和中等电压迟滞。基于操作同步加速器XRD,XAS和EDS结果,我们已经确认Se和Ca之间的全电池电化学反应是可逆的。未来的研究将确定增加比容量、降低电压迟滞和提高循环性的条件。这项工作表明Se基电极是有前途的CIB正极,并将促进对其他硫族化合物作为CIBs正极材料的进一步研究。
审核编辑:刘清
-
阴极碳酸盐中碳酸钙含量的络合滴定测定 GB 10304.3-2010-04-26 924
-
熔融碳酸盐燃料电池2009-10-23 1634
-
钛基电极材料助力未来电池可持续设计2020-04-03 3326
-
美国研发压力辅助技术,可提高钙钛矿太阳能电池的效率2020-05-22 4098
-
利用UV-NIR超快激光诱导磷硼酸盐玻璃来合成CsPbBr3量子点2020-08-06 1290
-
钙钛矿薄膜电池:成本仅为硅基电池的十分之一2020-12-09 5415
-
美国阿贡国家实验室John T. Vaughey:硼酸盐基可充钙硒电池2023-04-19 1726
-
可弯曲的未来能源:钙钛矿太阳能电池的新领域2023-11-06 2290
-
隆基晶硅-钙钛矿叠层电池效率达到33.9%2023-11-15 1731
-
科普知识丨热重分析仪TGA测试蔡基蒽硼酸水分和有机物含量2024-11-07 981
-
湿热与光老化条件下,封装工艺对碳基钙钛矿电池降解机理的影响2025-04-18 1615
-
隆基再次刷新晶硅-钙钛矿叠层电池转换效率世界纪录2025-04-27 1379
-
混合沉积法制备效率26.46%的钙钛矿/有机叠层电池及其稳定性研究2025-09-19 1290
全部0条评论

快来发表一下你的评论吧 !
