

H掺杂Ga2O3的缺陷计算(准备计算PREPARE02)
描述
DASP (Defect and Dopant ab-initio Simulation Package)是一款半导体缺陷和杂质性质的第一性原理计算模拟软件包,该软件包能针对输入的半导体晶体结构,基于材料基因组数据库和第一性原理软件包,自动计算并输出半导体的热力学稳定性,缺陷和杂质形成能及离化能级,半导体样品中缺陷、杂质和载流子浓度及费米能级,关键缺陷和杂质诱导的光致发光谱、载流子辐射和非辐射俘获截面及少子寿命。
针对任一半导体,DASP软件可以计算给出如下性质:热力学稳定性、元素化学势空间的稳定范围、缺陷(含杂质,下同)形成能、缺陷转变能级、各生长条件下的费米能级、载流子和缺陷浓度、缺陷的光致发光谱、缺陷对载流子的俘获截面、辐射和非辐射复合速率等。
本期将给大家介绍DASP HfO2的本征缺陷计算 5.3.1.2-5.3.1.3 的内容。
5.3.1.2. 使用DASP产生必要输入文件
新建目录doping-Ga2O3,在./doping-Ga2O3/目录内同时准备好以上的 POSCAR 文件与 dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。
5.3.1.3. PREPARE模块运行流程
产生超胞:
首先程序将根据min_atom=200和max_atom=250的参数,自动寻找最优的扩胞方案(即尽量使a=b=c且a⊥b⊥c),并给出超胞的POSCAR文件。以下为Ga2O3结构的超胞 POSCAR_nearlycube :
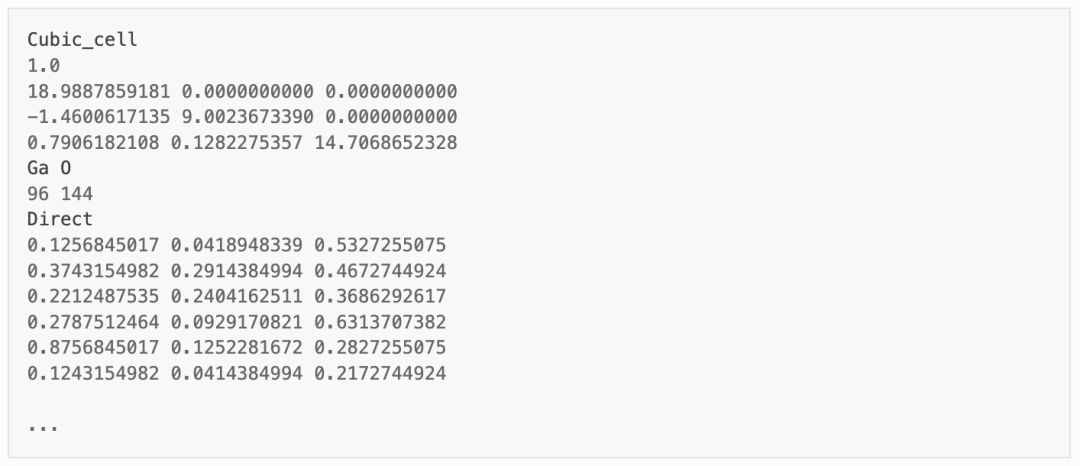
将其拖入晶体可视化软件,如图所示。
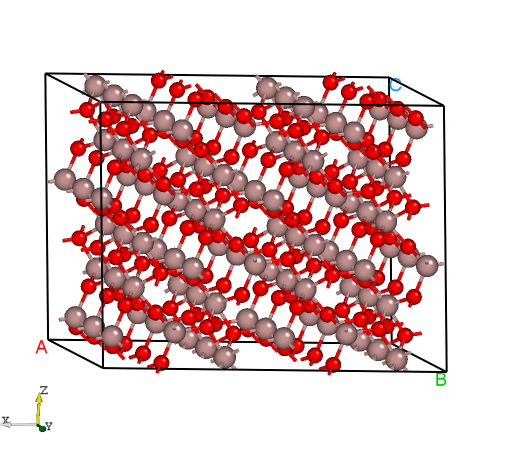
DASP产生的Ga2O3超胞的晶体结构。
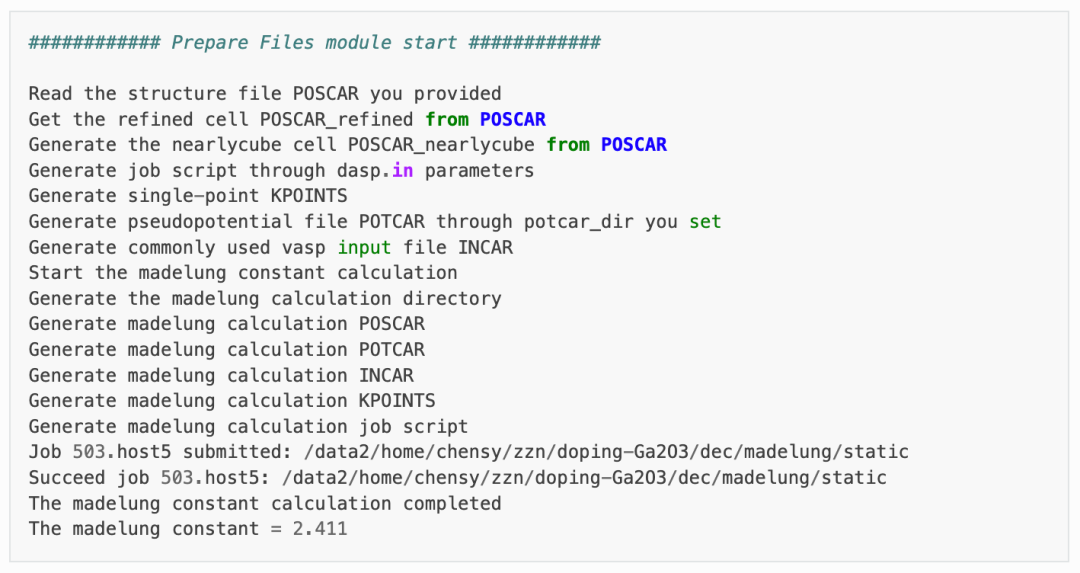
马德隆常数计算:
随后程序将根据产生的超胞文件,执行马德隆常数的计算,用来描述点电荷与均匀背景电荷的库伦相互作用。(用于Lany-Zunger修正)
以上两步计算完成,可观察 1prepare.out 的输出如下:
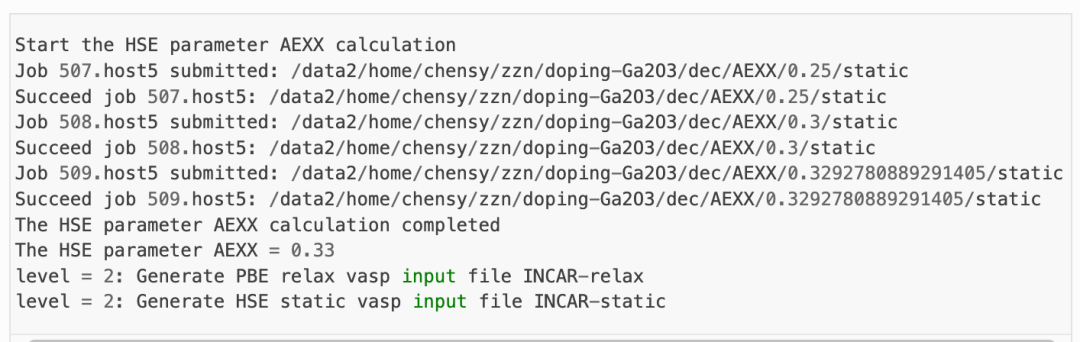
HSE交换参数计算:
程序将根据产生的超胞文件,先做AEXX=0.25和AEXX=0.3的HSE静态计算,从而根据斜率确定匹配 Eg_real = 4.9 的AEXX值,若AEXX=0.25或AEXX=0.3时带隙值与设置参数一致,则不会进行后续AEXX计算。因此,待计算完成后,可见doping-Ga2O3/dec/AEXX/目录内如下:
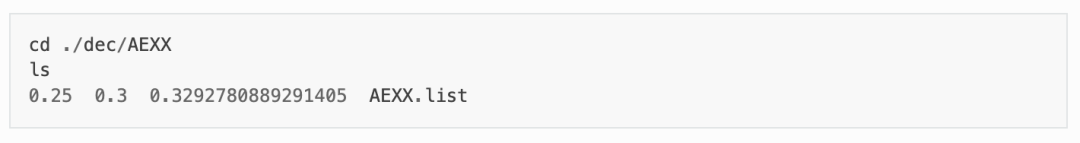
这表明当AEXX = 0.33(保留两位小数)时,Ga2O3超胞的带隙值为4.9 eV,将参数写入INCAR。同时从 1prepare.out 可以看到如下日志:
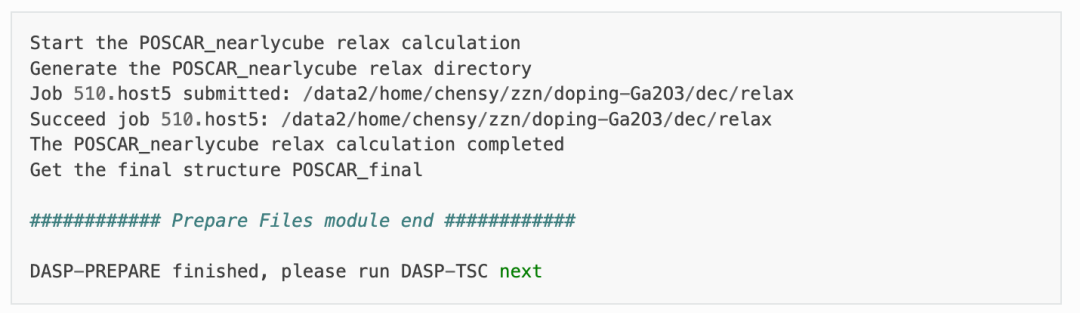
host超胞原子位置的优化:
PREPARE模块最后一步将根据level=2(即PBE优化)优化超胞内所有的原子位置,并在dec目录下产生最终的结构文件 POSCAR_final 。优化计算可见doping-Ga2O3/dec/relax目录。同时也可以在 1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算。
审核编辑 :李倩
-
镁掺杂对In2O3电导和气敏性能的影响2009-07-14 705
-
Ga2O3-SBD计算模型为分析器件物理提供了重要的参考价值2020-11-02 1210
-
最新成果展示:Ga2O3材料数据库的开发及其在日盲紫外光电探测器中的应用2022-10-25 2562
-
H掺杂Ga2O3的缺陷计算(准备计算PREPARE01)2023-04-24 2123
-
H掺杂Ga2O3的缺陷计算(热力学稳定性和元素化学势计算TSC)2023-04-26 1786
-
H掺杂Ga2O3的缺陷计算(缺陷形成能和转变能级计算DEC)2023-04-27 4213
-
ZnGeP2的本征缺陷计算之准PREPARE2023-05-19 1420
-
氧化镓(Ga2O3)沟槽二极管的相关研究进展2023-06-21 2197
-
中山大学王钢教授团队在NiO/β-Ga₂O₃异质结在功率器件领域的研究进展2023-06-30 3468
-
Ni掺杂β-Ga2O3单晶的光、电特性研究2023-10-11 3299
-
基于交变栅压调制的Ga2O3光电探测器2023-12-05 916
-
一种用于调控Ga2O3薄膜的表面电子结构的的热重组工程2024-01-19 3044
-
上海光机所在n型β-Ga2O3单晶光电性能调控方面取得进展2025-02-28 1135
全部0条评论

快来发表一下你的评论吧 !
