

原位紫外-可见光谱监测电池中氧化还原过程
描述
01
导读
为了满足对功率和能量密度的不同要求,开发使用各种电荷存储机制的电化学储能技术至关重要。电荷存储机制大致分为三个主要类型:电池型氧化还原、赝电容和电双层(EDL)存储。电池型电荷储存通常是由扩散或成核控制的法拉第过程,伴随着电子转移和相变。电双层电容器(EDLCs)通过离子在电极材料表面的非法拉第式电吸附来储存能量,这导致快速充电和高功率。赝电容存储的特点是表面控制的电荷存储,其能量密度比EDLC高,功率密度比电池大。与这三种机制相关的一个重要问题是:如何有效区分特定电极-电解质系统的电荷储存机制?由于电荷储存过程中电极材料的成分/结构变化的多样性和复杂性,先进的原位或非原位表征技术,如透射电子显微镜(TEM)中的电子能量损失谱(EELS),中子和X射线散射,光学显微镜和振动光谱学,被用来了解电荷储存机制。然而,所有上述技术都有局限性,包括仪器成本和可及性。
02
成果简介
该工作以原位紫外线-可见光(UV-Vis)光谱方法来区分电池型、赝电容和电双层电荷存储过程。相关工作以“In situ monitoring redox processes in energy storage using UV–Vis spectroscopy”为题发表在Nature Energy期刊上。
03
关键创新
1、使用价格低廉、易于获得、高速且无破坏性的紫外-可见光谱法监测电化学系统中氧化还原活动(图1);
2、使用多电位步长计时法(MUSCA)和CV方法与原位紫外-可见光谱法相结合来确定能量储存机制。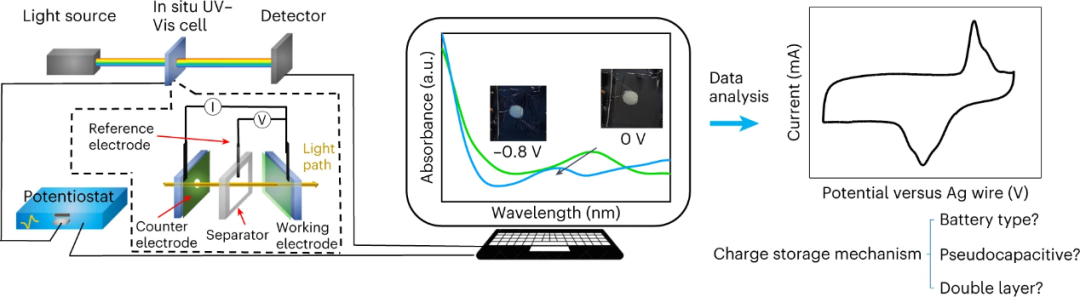
04
核心内容解读
1、原位紫外可见光电池的电化学特性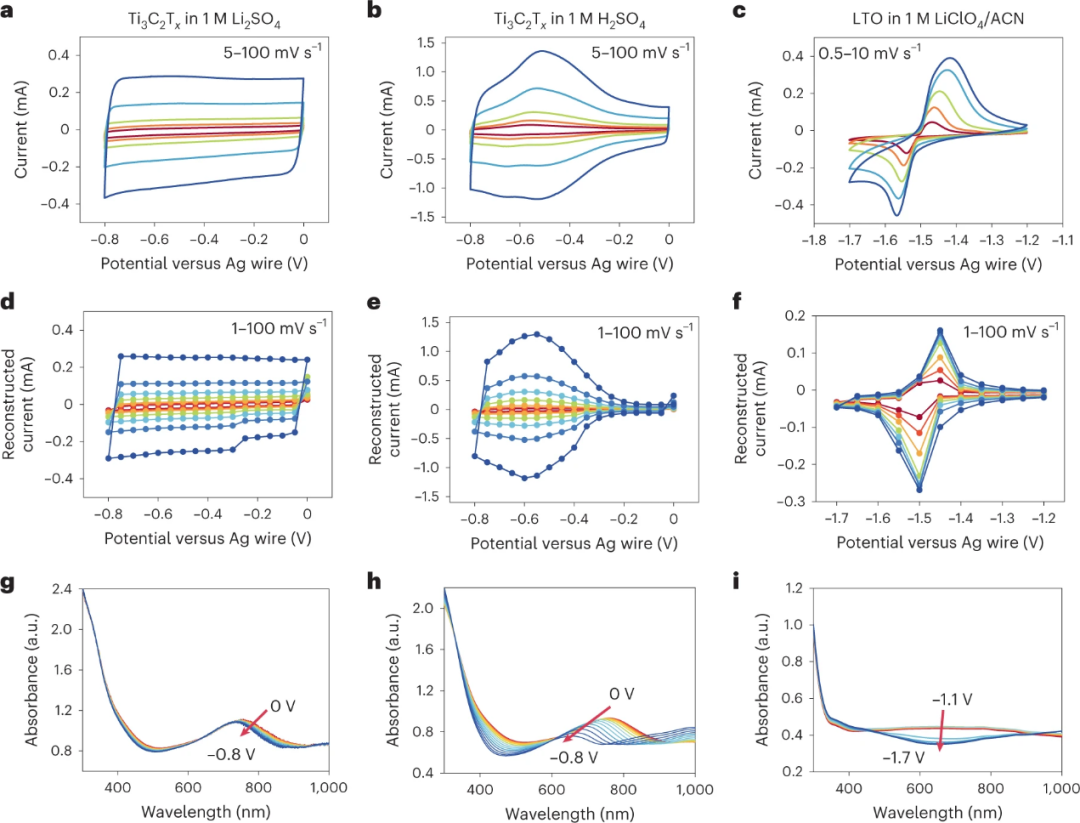
图 2. CV曲线,重建的CV曲线和原位电化学UV-Vis光谱。a-c,原位UV-Vis电池在不同扫描速率下的CV曲线。d-f,根据MUSCA数据重建的CV曲线。g-i,在阴极循环中的原位紫外可见吸收光谱。
CV被认为是确定电极的主导电荷储存机制的实用方法。在图2a-c所示的原位UV-Vis电池中对上述电化学系统进行了CV。Ti3C2Tx在1M Li2SO4中的CV曲线在扫描速率为5至100 mV s-1时几乎是矩形的(图2a)。同时,在1M H2SO4中的Ti3C2Tx在CV曲线的阳极和阴极分支上显示了一对宽峰,相对于Ag的电压为~-0.5 V(图2b)。
在1M Li2SO4系统中的Ti3C2Tx是由EDL形成主导的,而在1M H2SO4系统中的Ti3C2Tx,质子化导致电荷转移。在1M LiClO4/ACN中的LTO显示出典型的电池型CV曲线,其中有一对尖锐的山峰,且分离度很大(图2c)。CV曲线中的尖峰对应于核控制的Faradaic反应,该反应涉及Li+(去)插层期间的相变。
为了获得紫外-可见光范围内的全部吸收光谱,首先使用MUSCA方法来建立电化学行为和原位紫外-可见光光谱之间的相关性。MUSCA允许重建图2d-f所示的电位为50 mV的CV曲线。阴极循环期间的光谱显示在图2g-i中,显示出良好的可逆性。
当从0 V扫描到-0.8 V时,Ti3C2Tx电极在1M Li2SO4中的质子峰显示出从770 nm下移到740 nm。同时,在1M H2SO4中的Ti3C2Tx的UV-Vis光谱在相同的电位窗口中,质子峰的下移幅度大得多,达到约65 nm。质子共振峰在酸性水电解质中更明显的移动是由于Ti3C2Tx的表面端点的质子化,一个赝电容过程。
2、区分电荷储存机制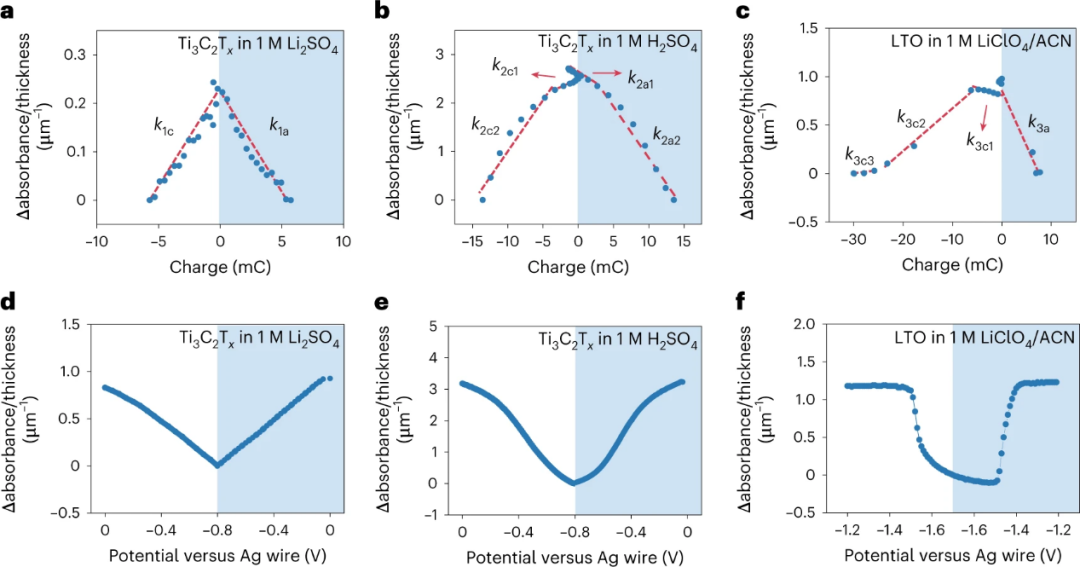
图 3. 原位UV-Vis光谱和电化学结果之间的相关性。a-c,用MUSCA方法计算Ti3C2Tx在1M Li2SO4(a),1M H2SO4(b)和钛酸锂(LTO)在1M LiClO4/ACN电解质(c)中的相对吸光度变化与电荷的关系。d-f,使用CV方法记录的1M Li2SO4中的Ti3C2Tx(d)、1M H2SO4中的Ti3C2Tx(e)和1M LiClO4/ACN电解质中的LTO(f)的相对吸光度变化与电位关系。
为了量化吸光度和电荷储存之间的关系,在图3中绘制了这三个系统的相对吸光度变化与电荷的关系。对于1M Li2SO4中的Ti3C2Tx,吸光度随电荷的变化几乎是线性的,而且在阴极和阳极循环之间没有区别(图3a)。对于1M H2SO4中的Ti3C2Tx,有两个线性斜率,分别对应EDL和赝电容充电。k2c1和k2a1代表在相对宽的电位范围内的两个EDL充电过程,而k2a2和k2c2代表赝电容的表面氧化还原反应。对于1M LiClO4/ACN中的LTO,有三个斜坡阶段,在阴极循环中尤其明显(图3c)。
总的来说,在所有三个系统中,相对吸光度的变化与不同电化学反应阶段内的电荷有良好的相关性。 通过分析相对吸光度变化与电位图的斜率和计算导数比,可以区分电池型、表面氧化还原和EDL电荷储存过程的区别。计算相对吸光度变化可以消除厚度和电位的影响(图3d-f)。对于在1M Li2SO4中以EDL为主的Ti3C2Tx的情况,从-0.8 V到0 V得到一个几乎线性的相对吸光度变化,斜率为-1.06 V-1 µm-1,如图3d所示。在1M H2SO4中的Ti3C2Tx在表面氧化还原反应的电位下也可以观察到一个更明显的相对吸光度变化。在-0.50 V时的斜率是-5.78 V-1 µm-1,这比EDL系统大五倍左右。在1M LiClO4/ACN系统中,围绕CV峰值位置-1.52 V计算出的LTO斜率为-16.22 V-1 µm-1,这比赝电容系统中的斜率大三倍,因为表面氧化还原反应发生在一个较窄的电位范围内。
3、电荷转移和紫外-可见光谱变化之间的耦合作用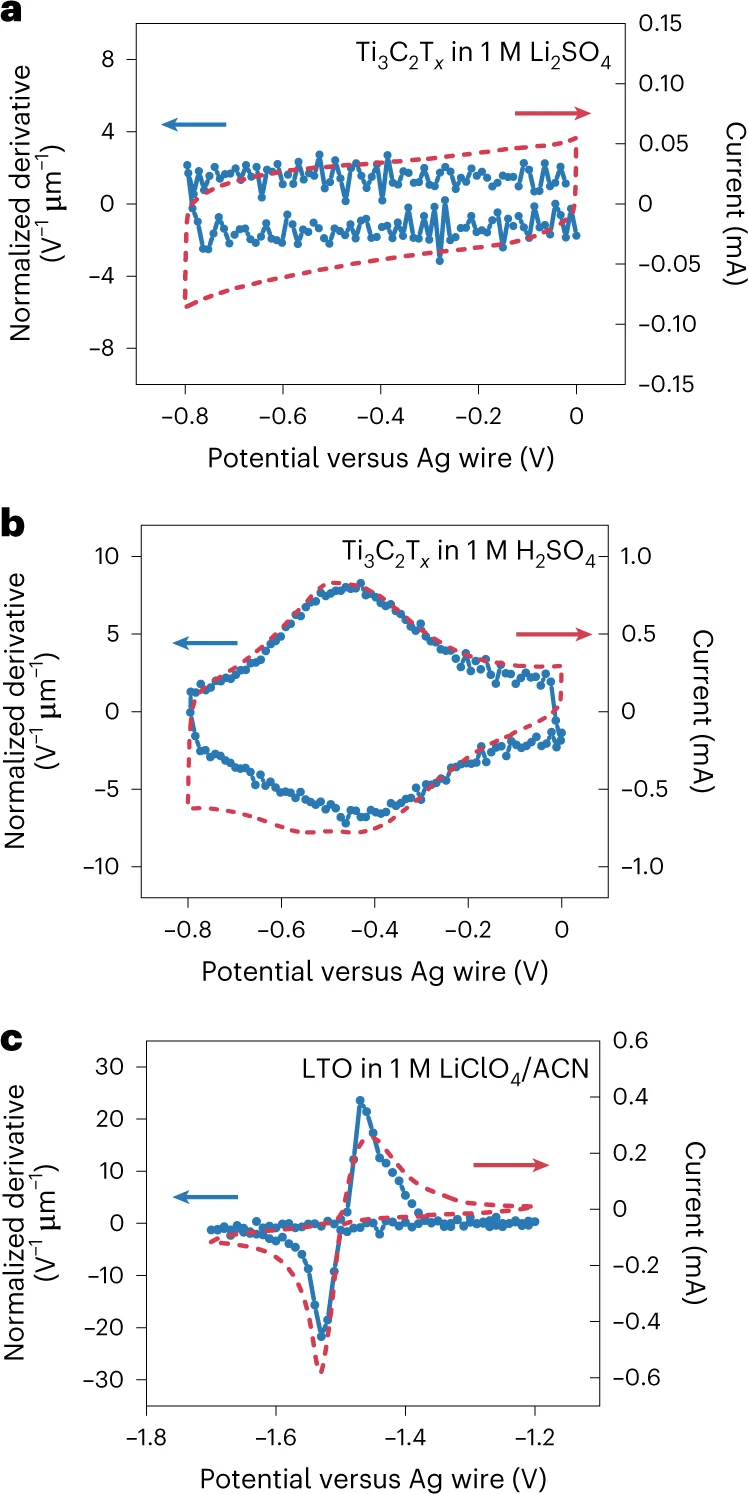
图 4.选定电化学系统的电化学和UV-Vis曲线的比较。a, Ti3C2Tx在1 M Li2SO4中10 mV s-1的电化学(红色)和UV-Vis CV(蓝色)曲线;b, Ti3C2Tx在1 M H2SO4中10 mV s-1的电化学(红色)和UV-Vis CV(蓝色)曲线;c, 钛酸锂(LTO)在1 M LiClO4/ACN中1 mV s-1的电化学(红)和UV-Vis CV(蓝)曲线。用于UV-Vis CV曲线的特征波长是Ti3C2Tx的450纳米和LTO的650纳米。
图4a-c用蓝色绘制了精度较高(每10 mV)的紫外可见光CV曲线。CV方法得出的UV-Vis CV曲线在形状、峰的起始电位和峰电位方面与它们相应的电化学CV曲线(图4-c)非常相似。紫外-可见曲线和电化学曲线之间的匹配说明电极材料的紫外-可见吸光度的变化对电荷状态的变化有高度的反应,因此可以广泛适用于在电荷存储时经历电子结构变化的材料,例如导电聚合物和陶瓷。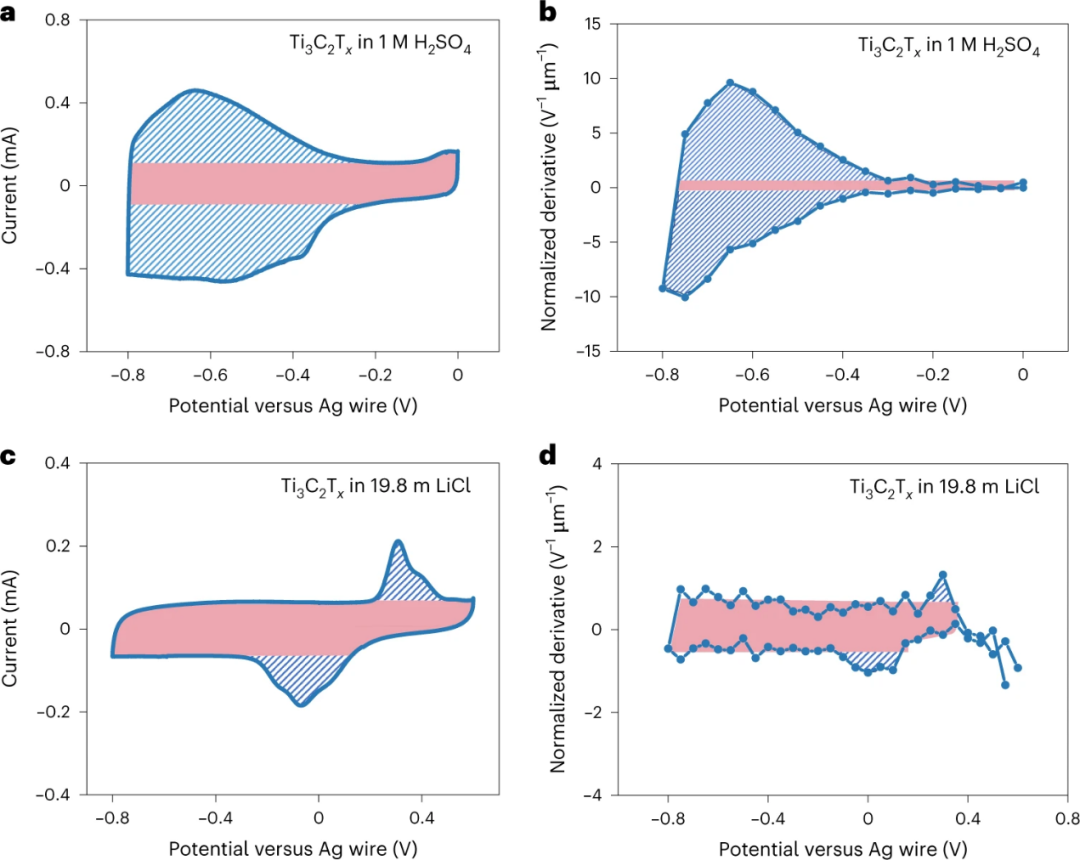
图 5. 不同电解质中Ti3C2Tx的EDL和表面氧化还原对总电荷的贡献。a,c, Ti3C2Tx在1 M H2SO4(a)和Ti3C2Tx在19.8 m LiCl(c)中的20 mV s-1的CV曲线。b,d, 用MUSCA方法收集的Ti3C2Tx在1 M H2SO4(b)和19.8 m LiCl(d)中的归一化导数与电位关系。粉红色区域表示在充电过程中EDL的贡献。
作者进一步比较Ti3C2Tx在不同电解质中的电流和归一化导数值,以确定Ti3C2Tx在19.8m LiCl中的电荷储存机制。通过比较阴影区的吸光度变化与EDL贡献的吸光度变化之比来确定电荷储存机制。
Ti3C2Tx在1M H2SO4中的平均氧化还原贡献的归一化衍生物是5.54 V-1 µm-1,大于EDL贡献的归一化衍生物0.58 V-1 µm-1,这证实了氧化还原贡献的归一化衍生物远远多于EDL贡献的归一化衍生物。另一方面,Ti3C2Tx在19.8 m LiCl中的蓝色阴影区域的平均归一化导数为0.25 V-1 µm-1,小于EDL-贡献的归一化导数0.62 V-1 µm-1。这表明,图5d中蓝色阴影区域的未知机制与EDL机制相似,不太可能涉及氧化还原过程。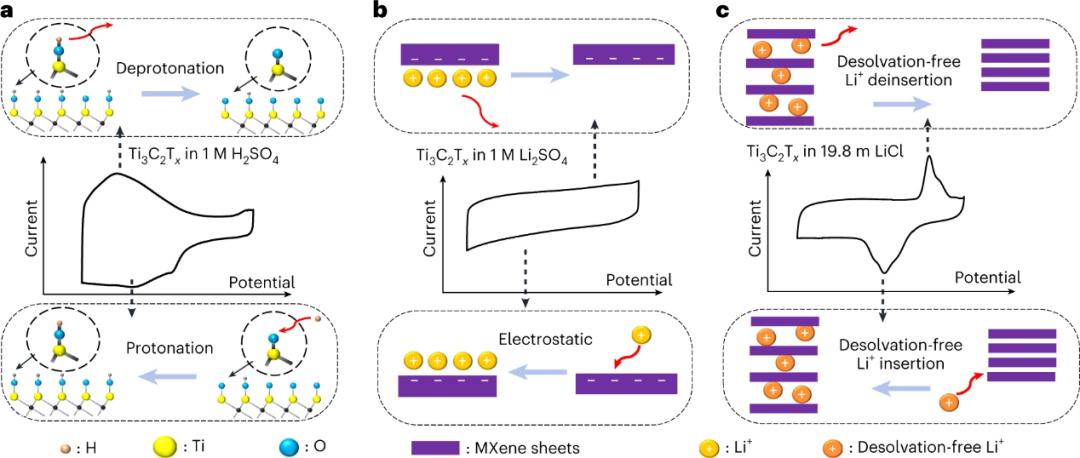
图 6. Ti3C2Tx在不同电解质中的电荷储存机制的示意图。a, 在阴极(底部)和阳极(顶部)过程中,质子化/去质子化主导了Ti3C2Tx在1M H2SO4中的电荷储存。c, 在阴极(底部)和阳极(顶部)过程中,无溶解的锂离子插入/剥离导致Ti3C2Tx在19.8 m LiCl中的CV曲线出现峰值。
Ti3C2Tx在不同电解质中的电荷储存机制主要有如下几种。如图6a所示,在1M H2SO4中的Ti3C2Tx的情况下,质子插层伴随着Ti的氧化状态变化。这是由于当插层的质子与Ti3C2Tx表面的=O基团结合时发生的表面氧化还原反应。在这种情况下,表面氧化还原反应有助于额外的电荷储存,导致在电化学CV曲线中观察到的峰值。另一方面,在1M Li2SO4中,没有氧化还原反应,插入的Li+在层间形成EDL,如图6b所示。因此,时间和电荷之间的线性关系形成一个矩形的CV曲线。如上所述,氧化状态的变化应导致紫外-可见光吸收光谱的明显变化。在这项工作中,原位紫外-可见研究的分析表明,非法拉第反应在这个过程中占主导地位,并消除了可能由电化学CV曲线中明确的峰的存在而引起的混淆。
05
成果启示
综上所述,本文证明了原位紫外-可见光谱是确定电荷储存机制和监测各种电化学系统中氧化还原过程的强大技术。利用紫外可见光的吸光度导数,显示了光谱变化和电化学过程之间的相关性,这种技术能够区分EDL、赝电容和基于插层的电池型氧化还原过程。由于其广泛的可及性和探测电子结构和颜色变化的独特能力,未来紫外光将在材料中广泛的电化学现象的研究中发挥越来越重要的作用,包括从能量储存到SEI形成、电解质分解、电催化、电致变色和材料性能的电化学调节。
审核编辑:刘清
-
中国科大研制可见光波段矢量光谱分析仪2025-08-28 953
-
太阳光谱全面解析丨UVA、UVB、UVC与可见光、红外光2025-07-24 9788
-
紫外可见光光度计使用步骤 紫外可见光光度计怎么用2024-02-18 16518
-
使用共阳极RGB LED来产生可见光谱的颜色2022-11-07 953
-
紫外吸收光谱仪概述及结构原理2022-04-19 9883
-
可见光通信 调制解调技术 家庭机器人 可见光通信应用 原理及硬件方案 精选资料分享2021-07-27 3739
-
可见光通信原理及硬件方案 精选资料分享2021-07-23 2819
-
GUVB-S31GD太阳光传感器韩国GENICOM紫外线可见光传感器2021-07-15 1532
-
紫外线传感器-GUVC-S30GD韩国GENICOM可见光传感器 光电二极管2021-07-13 827
-
求大佬分享一种多光谱可见光遥感图像压缩系统的设计方案2021-06-02 1410
-
如何利用可见光监视器2021-04-23 1188
-
合理利用不可见光照明提高视觉检测效率之紫外照明篇2018-06-14 2676
-
可见光照明连续消毒是什么?2018-06-07 5557
-
可见光音频发射机2009-09-22 914
全部0条评论

快来发表一下你的评论吧 !
