

双钙钛矿材料稳定性的快速计算预测(Cs2AgBiCl6 01)
描述
DASP (Defect and Dopant ab-initio Simulation Package)是一款半导体缺陷和杂质性质的第一性原理计算模拟软件包,该软件包能针对输入的半导体晶体结构,基于材料基因组数据库和第一性原理软件包,自动计算并输出半导体的热力学稳定性,缺陷和杂质形成能及离化能级,半导体样品中缺陷、杂质和载流子浓度及费米能级,关键缺陷和杂质诱导的光致发光谱、载流子辐射和非辐射俘获截面及少子寿命。
针对任一半导体,DASP软件可以计算给出如下性质:热力学稳定性、元素化学势空间的稳定范围、缺陷(含杂质,下同)形成能、缺陷转变能级、各生长条件下的费米能级、载流子和缺陷浓度、缺陷的光致发光谱、缺陷对载流子的俘获截面、辐射和非辐射复合速率等。
本期将给大家介绍DASP 双钙钛矿材料稳定性的快速计算预测 5.5-5.5.1.1 的内容。
5.5. 双钙钛矿材料稳定性的快速计算预测
在以上的案例中,我们展示了TSC模块可以计算元素化学势,用于DEC模块的缺陷形成能计算。此外,TSC也可以独立运行来分析目标化合物的稳定性,而无需先行做PREPARE模块的计算。
在 dasp.in 中设置 tsc_only = T ,可以直接运行TSC模块,待TSC结束第一阶段分析后,将自行做 level = 1 的第二阶段分析。
以下示例为 Cs2AgBiCl6 与 Rb2LiInI、 K2LiYF6 三种双钙钛矿材料的分析过程。
5.5.1. Cs2AgBiCl6 (预测结果:稳定)
5.5.1.1. 准备文件
利用 TSC 模块快速分析材料稳定性的第一步仍然是准备好 POSCAR 与 dasp.in 文件。
材料 Cs2AgBiCl6 的 POSCAR 文件可参考 Materials Project 数据库获得,需用户自行优化或设置晶体结构。本案例采取的 POSCAR 文件如下:
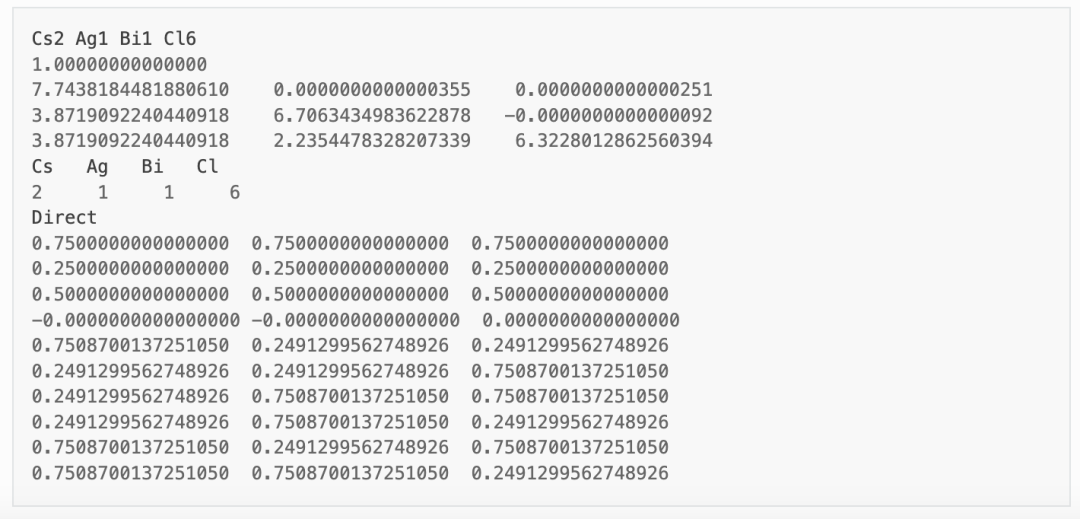
在 dasp.in 文件中,用户需根据自身情况设置任务脚本相关参数,并设置 tsc_only = T 以及 database_api 。
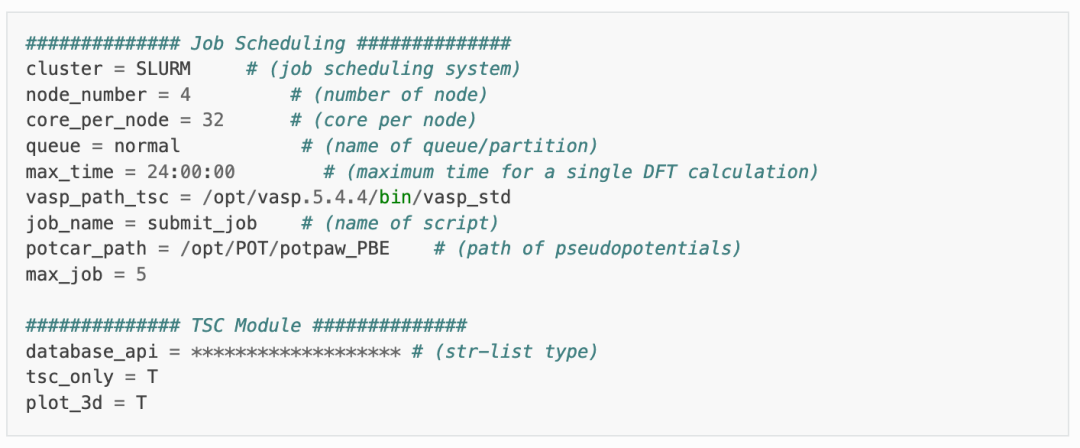
其中,对于 TSC 模块的参数:
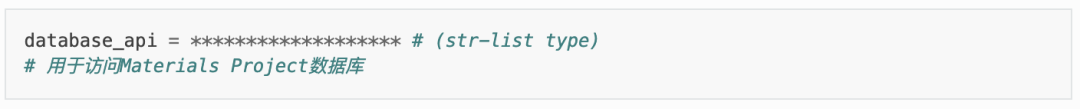
tsc_only = T # 仅进行 level = 1 的稳定性快速分析。
plot_3d = T # 对四元化合物,本模块可输出三维相图(仅供用户参考),该参数默认为F,设置为T即可输出三维相图。
审核编辑 :李倩
-
效率为25.1%的倒置钙钛矿太阳能电池中实现了高稳定性2025-11-14 378
-
钙钛矿电池的季节性效应:MPPT揭示衰减机制与稳定性优化2025-07-16 871
-
钙钛矿太阳能电池的降解机制和稳定化技术,解决实际应用中面临的稳定性问题2025-01-24 2107
-
接触角测量揭示TTC疏水层对钙钛矿太阳能电池稳定性的影响2024-11-27 1908
-
钙钛矿太阳能电池稳定性测试的最大功率点跟踪(MPPT)2024-10-10 2667
-
钙钛矿/晶硅叠层太阳能电池稳定性测试2024-07-25 3036
-
钙钛矿太阳能电池的湿热稳定性与效率优化2024-04-30 2078
-
DASP 双钙钛矿材料稳定性的快速计算预测2023-05-24 1182
-
双钙钛矿材料稳定性的快速计算预测(Cs2AgBiCl6 02)2023-05-16 1325
-
基于大数据的钙钛矿太阳能电池稳定性分析方法2023-01-12 3311
-
基于大数据将统计学方法在钙钛矿稳定性评估上的应用2022-12-23 1655
-
纯红光钙钛矿发光二极管提升光谱稳定性2022-11-22 1578
-
半导体所在高效稳定钙钛矿太阳能电池方面取得进展2022-07-31 1644
-
制备方法对Ba2FeMoO6双钙钛矿磁性能的影响2009-05-26 2478
全部0条评论

快来发表一下你的评论吧 !
