

ZnGeP2的本征缺陷计算之准PREPARE
描述
DASP (Defect and Dopant ab-initio Simulation Package)是一款半导体缺陷和杂质性质的第一性原理计算模拟软件包,该软件包能针对输入的半导体晶体结构,基于材料基因组数据库和第一性原理软件包,自动计算并输出半导体的热力学稳定性,缺陷和杂质形成能及离化能级,半导体样品中缺陷、杂质和载流子浓度及费米能级,关键缺陷和杂质诱导的光致发光谱、载流子辐射和非辐射俘获截面及少子寿命。
针对任一半导体,DASP软件可以计算给出如下性质:热力学稳定性、元素化学势空间的稳定范围、缺陷(含杂质,下同)形成能、缺陷转变能级、各生长条件下的费米能级、载流子和缺陷浓度、缺陷的光致发光谱、缺陷对载流子的俘获截面、辐射和非辐射复合速率等。
本期将给大家介绍DASP ZnGeP2的本征缺陷计算 5.4-5.4.1.1 的内容。
5.4. ZnGeP2的本征缺陷计算
ZnGeP2是一种非线性光学材料,但是其带隙内存在的较多光吸收峰限制了其应用,实验上认为这些吸收与点缺陷相关。因此,有必要对ZnGeP2的点缺陷性质开展理论计算,分析不同制备环境下其吸收峰的来源。
以下开始为使用DASP软件包计算ZnGeP2本征点缺陷的实例:
5.4.1. 准备计算PREPARE
5.4.1.1. 准备POSCAR与dasp.in
获得ZnGeP2材料的结构文件 POSCAR,使用VASP优化其晶格常数,或修改其晶格常数从而匹配实验值(此步骤由用户自行完成)。显示如下:
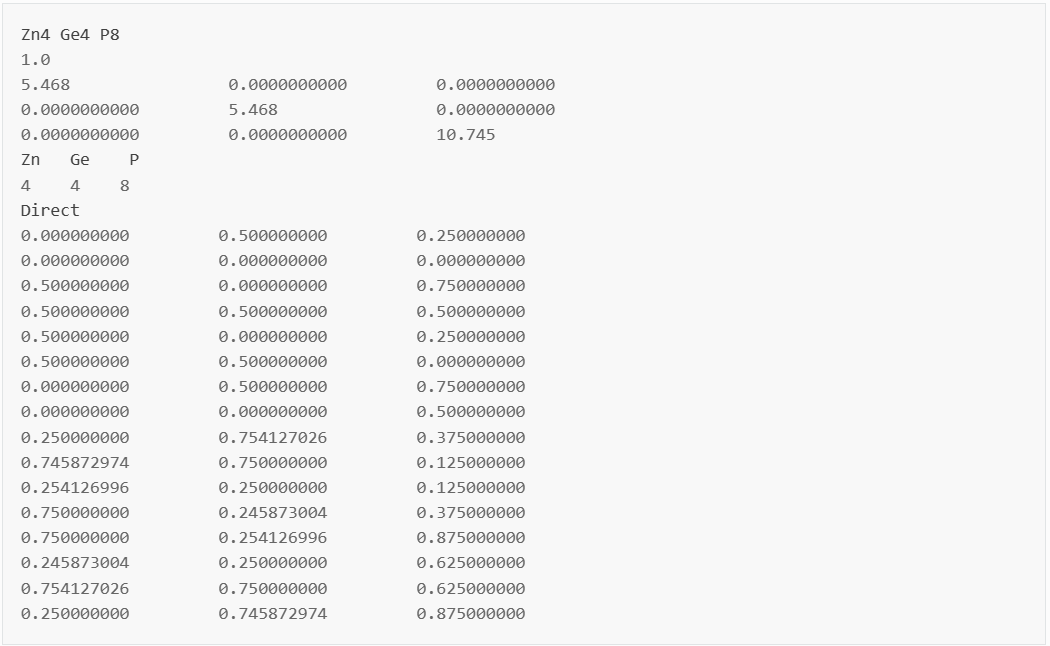
在晶体可视化软件中如图1所示。

ZnGeP2的晶体结构。
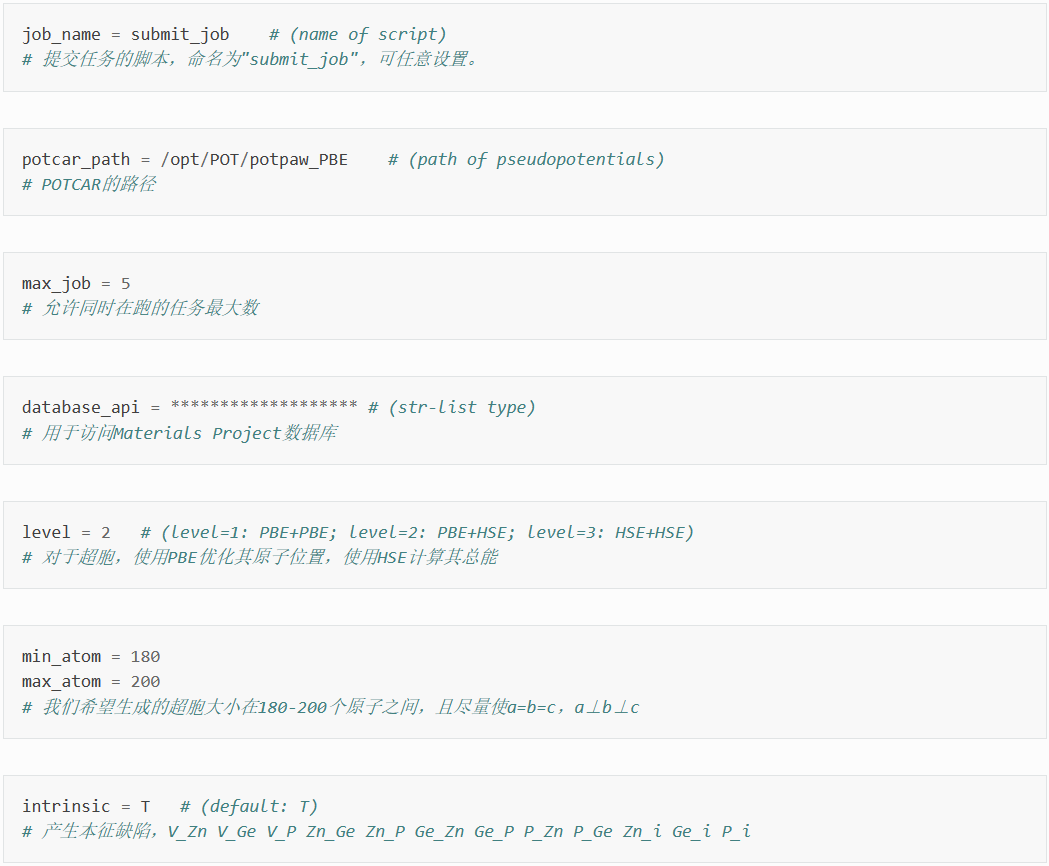
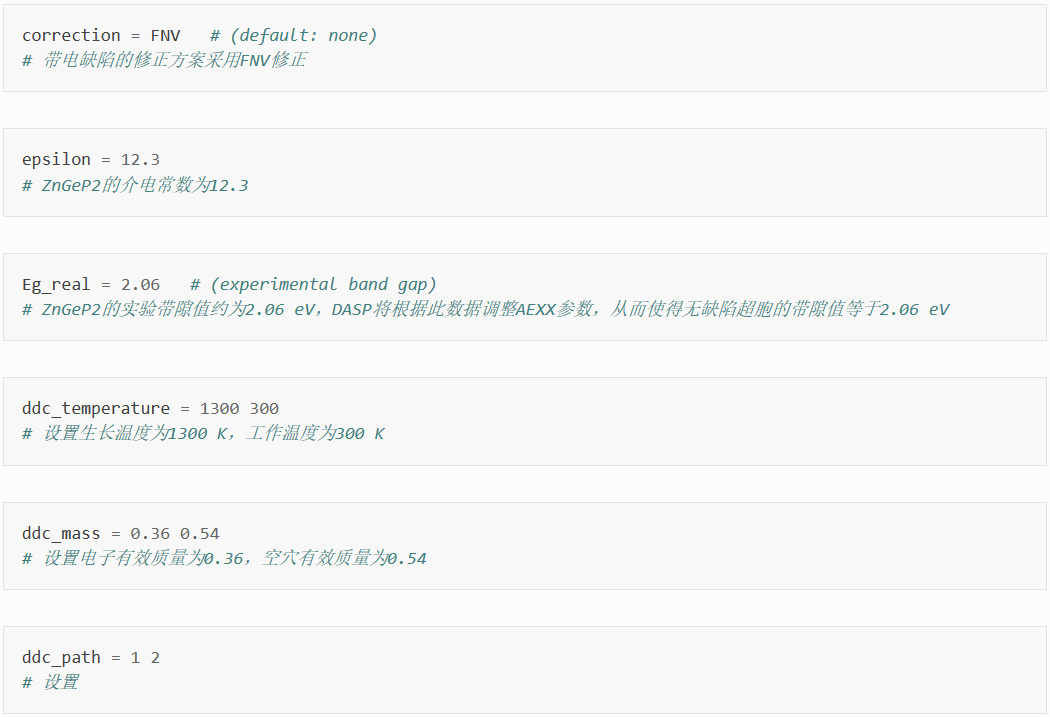
在dasp.in中写入必要参数

接下来将对dasp.in中所有列出的参数进行说明。



编辑:黄飞
-
ZnGeP2的本征缺陷计算之PREPARE模块运行流程2023-05-19 1271
-
基于DASP ZnGeP2的本征缺陷计算2023-05-16 1447
-
ZnGeP2的本征缺陷计算(非辐射俘获系数计算CDC)2023-05-11 1827
-
半导体缺陷原理:DASP HfO2的本征缺陷计算2023-04-24 2667
-
DASP HfO2的本征缺陷计算(缺陷形成能和转变能级计算DEC)2023-04-18 4396
-
一文解析DASP CdTe的本征缺陷计算2023-04-05 1510
-
5.3.1.1 本征缺陷∈《碳化硅技术基本原理——生长、表征、器件和应用》2022-01-06 1648
全部0条评论

快来发表一下你的评论吧 !
