

NTO表面和分子空位对其热反应动力学的影响
描述
0 1 引言 NTO作为一种新型的火炸药,其爆速和爆压与HMX相近,对冲击、火花、热和冲击的敏感性低于RDX和HMX,近几十年来备受关注。国内外对NTO单分子的热分解机理进行了大量的实验或理论研究,但考虑到NTO晶体中分子间的相互作用,其分解机制仍不清楚,可能不同于单分子的分解机制。由于材料固有的物理化学性质和工业技术的微小缺陷,合成的NTO晶体存在晶面和分子空位。相关研究表明,空位和表面效应在一些含能材料的起爆过程发挥重要作用。因此,我们通过第一性原理计算研究了NTO表面和分子空位对其热反应动力学的影响。 0 2 成果简介
在我们的工作中,基于自洽电荷密度泛函紧束缚(SCC-DFTB)的分子动力学模拟,研究了NTO晶体的热分解机制,并研究了表面和分子空位对其热稳定性的影响。观察到最初的分解反应主要是分子内化学键的断裂,并提出了三个主要的反应途径:硝基的裂解、环内C3-N2键的断裂和H原子转移到羰基,其中C5 - N5键断裂在高温下起主导作用。在分子空位和表面,我们发现硝基基团裂解和H原子转移的反应势垒都低于理想的NTO晶体,表明它们可以对NTO晶体的热稳定性和分解反应动力学有效的调控。我们的工作可能对设计具有更好的性能、安全性和可用性的高能量密度材料有所启发。
0 3 图文导读
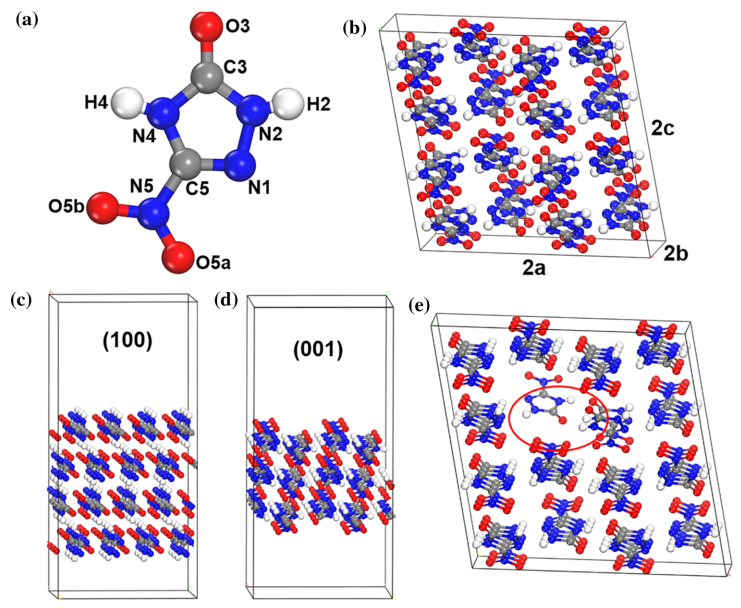
图1. (a) NTO分子结构 (b) NTO的2×2×2超胞 (c) NTO的(100)和(001)表面 (d)具有三个分子空位的NTO超胞
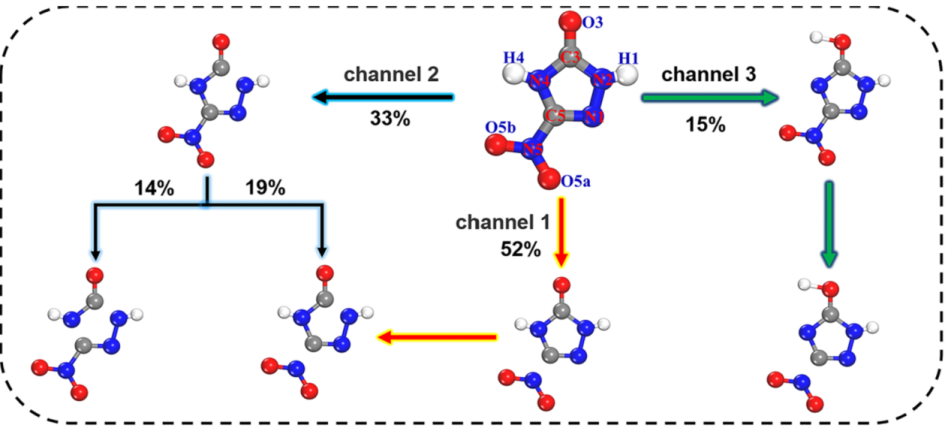
图2. NTO晶体初始热分解反应阶段的主要反应通道
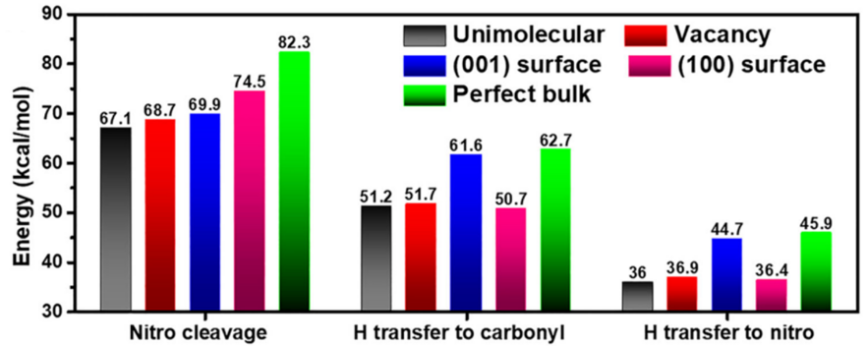
图3. DFT计算不同NTO结构的分解反应势垒
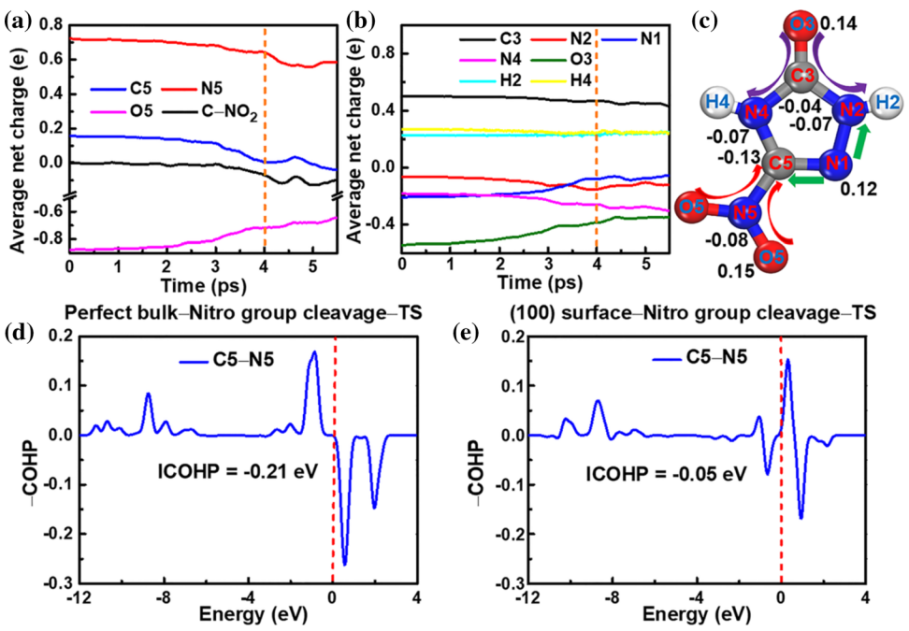
图4. 电子结构性质分析。初始分解过程中 (a) C‒NO2基团和 (b) 其它原子的平均净电荷演化。(c) 电荷转移演化原理图。(d) 理想晶体和 (e) (100)表面NTO分子的硝基断裂反应过渡态结构C5‒H5化学键的COHP
0 4 小结 在我们的工作中,利用鸿之微DS-PAW软件进行分子动力学模拟,得到基于从头算的轨迹结构以及对应的能量、原子受力等信息。基于自洽电荷密度泛函紧束缚(SCC-DFTB)的分子动力学模拟,研究了NTO晶体的热分解机制,并研究了表面和分子空位对其热稳定性的影响。观察到最初的分解反应主要是分子内化学键的断裂,并提出了三个主要的反应途径:硝基的裂解、环内C3 - N2键的断裂和H原子转移到羰基,其中C5-N5键断裂在高温下起主导作用。对于分子空位和表面结构,我们发现其硝基基团裂解和H原子转移的活化势垒低于理想的NTO晶体,表明它们可以对NTO晶体的热稳定性和分解反应动力学有效的调控。此外,基于DFT的电子结构计算验证了上述反应机理以及分子空位和表面的影响。
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 577
-
电力拖动系统的动力学课件2008-11-19 4516
-
[下载]想了解多体动力学软件吗?有教程分享及免费试用下载2009-03-24 4393
-
基于多体系统动力学的空气悬架大客车平顺性试验仿真研究2009-12-02 4154
-
汽车系统动力学长篇大论2017-11-01 5179
-
飞行器动力学参数在线辨识EKF算法实验流程2021-08-27 1499
-
分布式驱动电动汽车的动力学控制有哪几种类型?常见问题是什么?2021-08-30 1442
-
热分析动力学2009-12-01 762
-
电动力学或经典电动力学统称为什么?2020-06-10 5220
-
拓展人类潜能:深势科技使用IPU赋能分子动力学2021-12-09 3442
-
硅晶片的化学刻蚀和动力学效应2022-04-15 1780
-
探讨高浓电解液阴离子对Li+活度和嵌入反应动力学影响机理2023-03-15 5937
-
使用分子动力学轨迹来预测大型自旋系统中的核自旋弛豫行为2023-05-20 2568
-
基于车辆动力学模型的横向控制2023-11-15 2359
-
超高灵敏度emICCD相机助力钻石中氮空位的量子动力学研究2023-12-25 1430
全部0条评论

快来发表一下你的评论吧 !
