

使用分子动力学轨迹来预测大型自旋系统中的核自旋弛豫行为
电子说
描述
分子动力学(MD)模拟对于研究小分子和大型生物分子系统的构象移动性非常有用,但对于MD力场预测的构象能量和分布的准确性仍存在疑虑。有令人鼓舞的迹象表明,MD可以合理预测构象之间的转变时间尺度,但这一成功必须得到验证:除了少数例外,力场参数通常设定为重现热力学性质和量子力学能量,而不是构象转变的速率或停留时间。随着开发出具有正确自扩散和粘度的新水模型,以及探索对蛋白质非结构区域建模的新方法,彻底的实验验证变得越来越重要。
一、写在文前
磁共振在探测构象及其动态特性方面具有独特的能力,尤其通过自旋弛豫和交叉弛豫测量。然而,大多数用于解释自旋弛豫数据的模型在局部移动性方面做出了简化的近似,并使用了孤立自旋对模型。最近出现了更为复杂的利用MD模拟进行NMR和EPR弛豫的方法,但它们仍然主要使用自旋对近似;我们小组的工作也属于这个类别。需要的是一个软件框架,能够将MD轨迹直接转换为任意自旋系统的弛豫超算符。
鉴于此,本文报告了在Spinach库中实现了这个功能。我们在使用OPC和TIP5P水的甘露糖(GLYCAM06力场)上测试了所提出的方法。甘露糖的构象动力学已经有了较好的研究,尤其是通过使用孤立自旋对近似解释的13C自旋弛豫测量。
在这里,我们将重点放在1H-1H核Overhauser效应(NOE)上,这种效应探测葡萄糖的H1环异构体质子(图1)和连接葡萄糖和果糖残基的糖苷键两侧的果糖H11,12质子之间的接触。我们证明,从1微秒长的MD轨迹中提取的(交叉)弛豫率与实验数据非常吻合,考虑到NOE差异为9-25%,这会转化为1.5-4.2%的距离误差。这对于GLYCAM06力场以及OPC和TIP5P水模型来说是一个好消息。核自旋(交叉)弛豫率很容易测量,我们建议将这样的测试纳入新型分子动力学力场的标准基准测试集中。相关工作以“Using molecular dynamics trajectories to predict nuclear spin relaxation behaviour in large spin systems”发表在《Journal of Magnetic Resonance》上。
二、图文速递
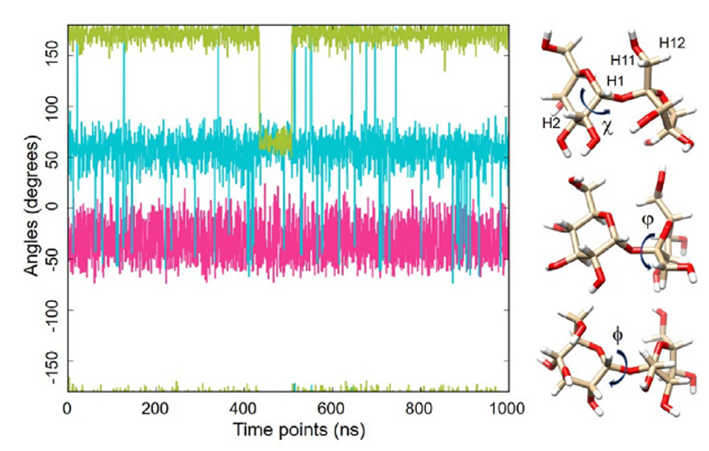
图1. 甘露糖内所示二面角的动力学特性(1毫秒MD轨迹,GLYCAM06甘露糖,OPC水,300K)。v(H2-C2-C1-O20)以绿色显示,u(C1-O20-C2-C1)以青色显示,/(H1-C1-O20-C2)以红色显示。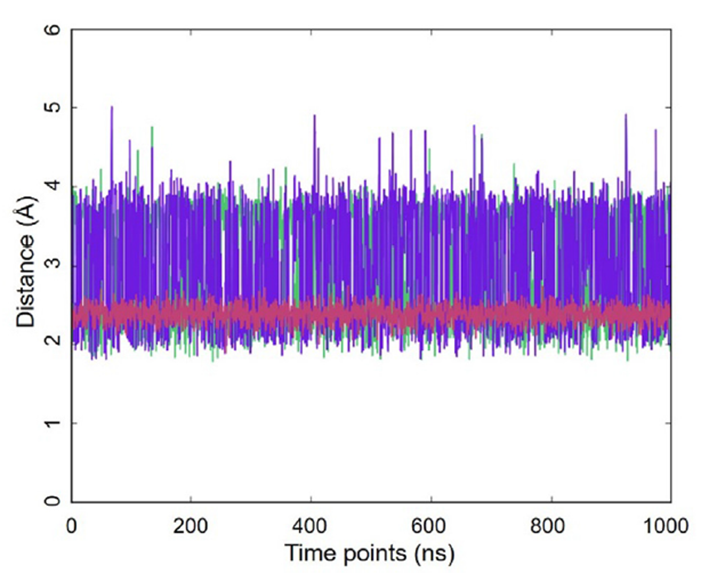
图2. 原子之间的距离动态(1毫秒MD轨迹,GLYCAM06甘露糖,OPC水,300K):葡萄糖H1到果糖H11的距离(绿色),葡萄糖H1到果糖H12的距离(紫色),葡萄糖H1到葡萄糖H2的距离(红色)。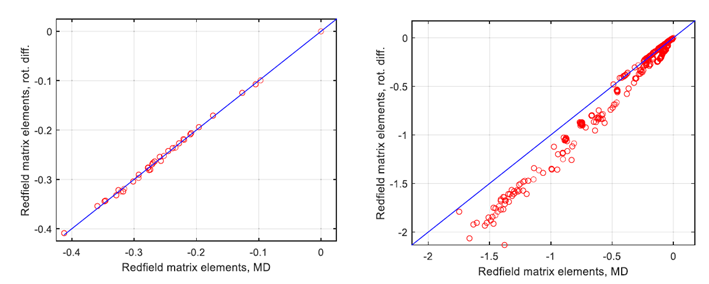
图3. 对比分析处理(使用纯旋转扩散近似)和数值处理(使用MD轨迹积分,OPC水)中弛豫超算符的非零元素之间的差异,随着内部运动对自旋弛豫的贡献增加。左侧面板:相对刚性的葡萄糖环中的3个自旋子系统(H1,H2,H4),其中偶极耦合调制几乎完全是旋转性质。(右侧面板)包含6个自旋子系统(葡萄糖的H1,H2,H3,H5;果糖的H11,H12),其中存在与糖苷键和外环C1-C2果糖键相关的显著内部运动。两个面板中的蓝线对应旋转相关时间为78皮秒,该值是通过拟合左侧面板的数据获得的。
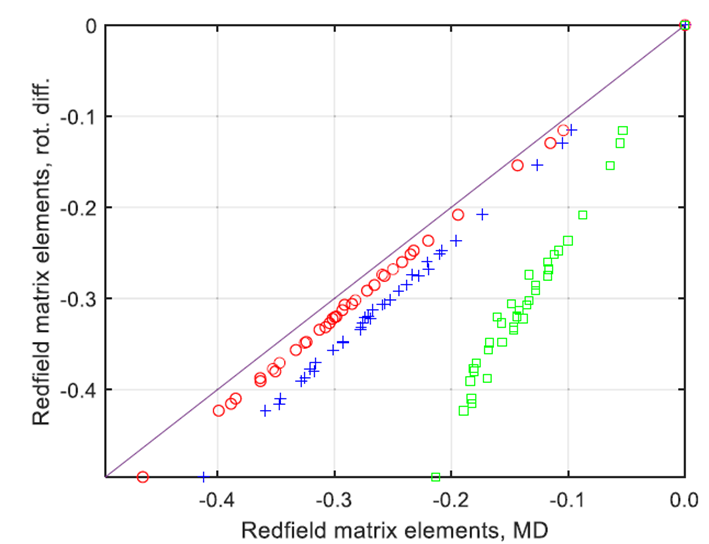
图4.来自TIP3P水(绿色方块)、TIP5P水(红色圆圈)和OPC水(蓝色十字)的分子动力学轨迹所得到的Redfield弛豫超算符的非零元素(X轴),与使用纯旋转扩散近似和旋转相关时间为100皮秒获得的相应元素的解析弛豫超算符进行对比(绿色方块代表TIP3P,红色圆圈代表TIP5P,蓝色十字代表OPC)。
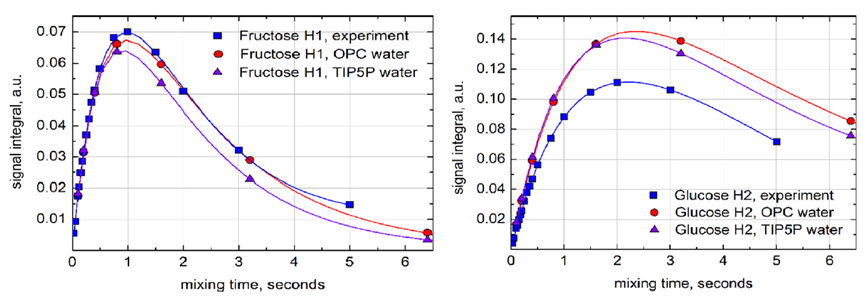
图5. 在葡萄糖H1质子自旋反转后,实验(蓝色)和模拟(OPC水为红色,TIP5P水为紫色)的核Overhauser效应(NOE)累积曲线的比较(曲线为双指数拟合)。模拟使用以葡萄糖H1为中心的8个自旋系统进行,包括(Glc H1-H5;Fruc H11,H12,H3)。信号积分是反转共振强度的分数。实验数据在600 MHz和308 K下收集;选择更高的温度是为了补偿D2O的增加粘度。没有尝试补偿蔗糖浓度对粘度的影响 - 预计该影响在0.5%以下。在进行核磁共振实验之前,样品被除气处理。
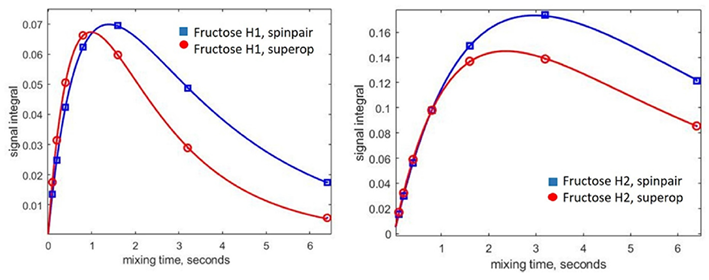
图6. 使用自旋对(蓝色)和完整自旋系统(红色)方法对模拟的核Overhauser效应(NOE)累积曲线进行比较(在葡萄糖H1共振反转时)。实线是双指数拟合。左侧面板:果糖H11 + H12质子。右侧面板:葡萄糖H2质子。
三、小结
我们提出了一个新工具,用于将实验自旋弛豫数据与基于分子动力学轨迹的模拟进行比较。它通过对Redfield积分进行数值评估直接计算自旋弛豫超算符。该工具已集成到Spinach软件包中,并能够模拟多种自旋弛豫实验,包括具有显著强耦合和弛豫干涉效应的自旋系统。这类计算直到最近才变得可行,其中有五个主要因素起到了贡献: (a)具备足够处理能力的图形卡的易得性(NVidia Ampere架构超过10TFLOPS的处理能力,约等于2002年全球最大超级计算机); (b)在磁共振中开发和实施了用于弛豫超算符和稀疏矩阵指数的大规模方法; (c)Matlab的性能和语法灵活性不断提高,其中的程序代码通常比用人类语言和标准数学符号描述更短且更易读; (d)出现了线性复杂度缩放的液态自旋动力学模拟方法,使得蛋白质大小的系统不再无法达到; (e)提供了足够长的分子动力学轨迹,以同时在方程中对上限和集平均值进行收敛。
我们通过在蔗糖上模拟NOE实验对该工具进行了测试,使用了GLYCAM06力场下的1毫秒分子动力学轨迹和OPC、TIP5P和TIP3P水模型。与实验的一致性非常好,考虑到0-25%的NOE初始斜率的差异对应刚性结构中的距离误差为0-4.2%。这对于GLYCAM06糖类参数化和OPC、TIP5P水模型来说是非常好的,但TIP3P效果不佳。上述模拟的CPU时间需求不超过运行分子动力学轨迹所需的时间。一旦计算了弛豫超算符,通过计算多个混合时间下的NOE或模拟任何其他弛豫实验所消耗的CPU时间是微不足道的。在具有24个CPU核心的系统中,使用IK-0(3)基组和每2皮秒采样一次的1皮秒轨迹,8自旋弛豫超算符计算大约需要30小时;如果使用GPU,这个时间将大大缩短。预计更大的自旋系统将变得简单:一旦自旋数超过受限态空间近似中的强相互作用团簇的大小(通常在液态NMR模拟中少于五个自旋),计算复杂性与自旋系统的大小大约呈线性关系。
审核编辑:刘清
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 556
-
Adams多体动力学仿真解决方案全面解析2025-04-17 8174
-
电力拖动系统的动力学课件2008-11-19 4497
-
基于多体系统动力学的空气悬架大客车平顺性试验仿真研究2009-12-02 4141
-
高梯度磁场中磁性微粒的动力学模型研究2010-05-13 2283
-
汽车系统动力学长篇大论2017-11-01 5149
-
什么是“量子自旋霍尔效应”?2018-12-13 5483
-
电子自旋与保利原理(续ii)2020-07-27 1928
-
热分析动力学2009-12-01 733
-
灰色预测模型的系统动力学仿真2011-08-01 1211
-
航天器姿态动力学与控制的详细资料说明2019-06-03 1843
-
激发态动力学第一性原理计算软件Hefei-NAMD2022-05-26 3729
-
NTO表面和分子空位对其热反应动力学的影响2023-05-19 4280
-
致真精密仪器自旋测试多功能克尔显微镜介绍2025-02-10 865
-
Nat. Mater.:室温下PdSe₂诱导的石墨烯平面内各向异性自旋动力学2025-02-17 1625
全部0条评论

快来发表一下你的评论吧 !
