

水系锌离子电池电解质设计原则
描述
01
导读
为了追求安全性和成本,人们开始关注水系电池。水系电解质有许多吸引人的优点,如不易燃和环保,但也有能量密度低的缺点。为了解决这一问题,人们开始重新配制水系电解质,但前提是要了解电解质的物理性质和电化学性能之间的相互作用。
02
成果简介
近期Materials Today期刊上发表了一篇题为“Electrolyte formulas of aqueous zinc ion battery: A physical difference with chemical consequences”的综述。该综述从电解液的组成出发,讨论了锌离子电池电解液是如何导致不同的电化学性能。通过对电池电化学性能的五个指标进行评价,为不同用途的电池提供了一种电解质设计策略。
03
核心内容解读
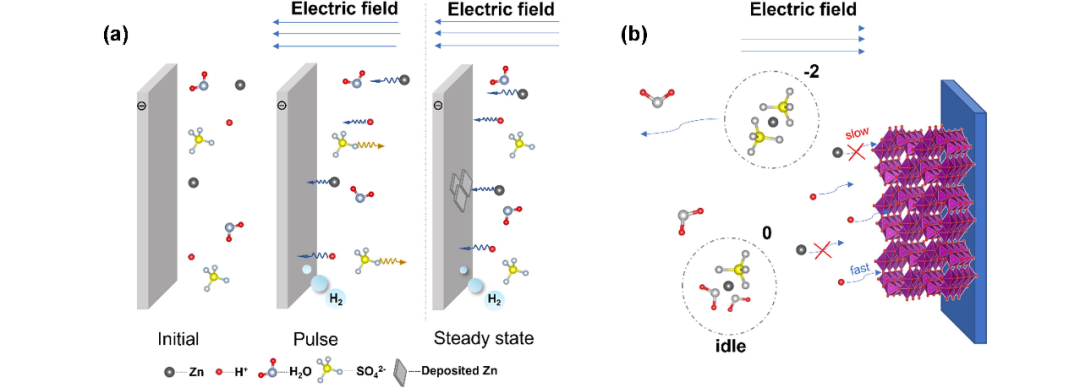
图1. (a)Bruce-Vincent法测量离子电导率示意图。(b)α-MnO2正极放电过程示意图。@Elsevier
电导率和转移数
电解液最重要的特性是能足够快地输送载流子。溶剂在电导率中起着重要的作用,这不仅是因为盐在特定溶剂中的浓度限制,还因为电荷载流子在溶剂化过程中在溶液中移动。为了量化离子在溶液中的输运能力,离子电导率(σ)表示为所有带电物质输运的总和。
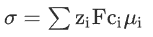
其中离子的电荷(zi)、浓度(ci)和迁移率(μi)影响离子电流的贡献。然而,对于摇椅型电池,溶液的高离子导电性并不能直接转化为良好的电化学性能,因为离子导电是所有离子物种(正电荷和负电荷)集体运动的结果,而法拉第电流仅由一种离子(大部分正电荷)平衡。为了解耦不同离子对离子传导的贡献,迁移数(ti)表示为
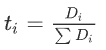
与溶液中其他离子相比,较高的ti对总离子电流的贡献更高,迁移率也相对较高。对于电池的工作,一个好的电解质不仅需要高离子电导率,还需要高载流子转移数,供应足够的载流子,以通过法拉第反应被氧化/还原。
在非水系电解质(碳酸盐或醚基)中,溶剂极化程度较低,难以移动,1M浓度可实现最大电导率(~10 mS/cm),提供足够的阳离子来运输,而不会增加电解质的粘度。由于非质子溶剂的简单性,Li+将是主要的正离子物种,其转移数为~0.4,比未溶剂化的阴离子略慢。
由于水作为溶剂的高介电常数,水系ZIB电池的电导率是非水系电池的10倍。典型的1 M ZnSO4水系电解质离子电导率为~50 mS/cm,然而,由于存在H+,这种高电导率可能被高估了,因为H+传导的跳跃机制也会贡献电流。增加Zn2+的浓度不仅会增加电解液的导电性,还会使溶液酸化,从而诱导更多的质子从水中排出,这些质子的跳跃机制极大地提高了离子的导电性,但对电池的运行不利。
显然,水溶液的高导电性是以Zn2+的低迁移数和H+的高迁移数为代价的。有没有办法在测量中区分它们?不幸的是,经典的Bruce-Vincent方法是基于锌是唯一氧化还原活性物质的假设,这是不正确的,因为H+/H(SHE, 0 V)的氧化还原电位高于Zn2+/Zn(-0.78 V vs. SHE)。更具体地说,它用稳态电流除以脉冲电流来计算传递数:
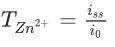
但稳态电流由Zn2+沉积和H+还原共同贡献,计算值为H+和Zn2+迁移数之和(图1a)。更糟糕的是,电解质浓度远远超出了Bruce-Vincent方法的准确度范围(低于0.01 M)。当ZnCl2和ZnSO4浓度超过~0.14 M时,Zn2+的转移数急剧下降。但在电解质中,这些离子对并不构成离子电流(负电荷离子对甚至抵消离子电流),如图1b所示。

图2. (a)不同浓度ZnCl2溶液的拉曼光谱。(b)沿[000](左)和[001](右)投影方向放电α-MnO2的扫描透射电镜(STEM)分析。(c)α-MnO2在第一次循环和第二次放电时的XRD。@Elsevier
飞秒受激拉曼光谱(FSRS)揭示了ZnCl2水溶液(5~30摩尔浓度(m))中离子的形态,作为离子对存在的有力证据(图2a),其中[Zn(OH2)2Cl4]在所有浓度中占主导地位,而Zn[(OH2)6]2+的强度要弱得多。在几种不同的溶液中也观察到这种正负离子形态。不幸的是,水系锌电池本质上总是会被离子对和质子纠缠,因为不仅Zn2+电导率的精确测量受到H+干扰,甚至ZIB正极的电化学性能也可能受H+(脱)插层的影响(图1b)。MnO2作为典型正极,其工作机理由于H+和Zn2+的共存而变得复杂。
在各种相中,α-MnO2首先以质子为MnO2→MnOOH转化反应的主要载流子,但后来这一机制与Zn2+插入和Zn2+/H+共插入的报道相矛盾,使其工作机制更加复杂。在H+转化、H+(脱)插嵌和Zn2+/H+共(脱)插嵌之间还没有达成一致。Lu等人最近提出利用高角度环形暗场(HAADF)分析放电后的α-MnO2,其缺少重离子的空通道支持H+的(脱)插嵌机制(图2b)。根据原位XRD, α-MnO2图案在所有荷电状态中都存在(图2c),因此排除了转换型机制。α-MnO2在硫酸锌电解液中复杂的工作机理归结为在保持隧道结构的同时(脱)插嵌H+。
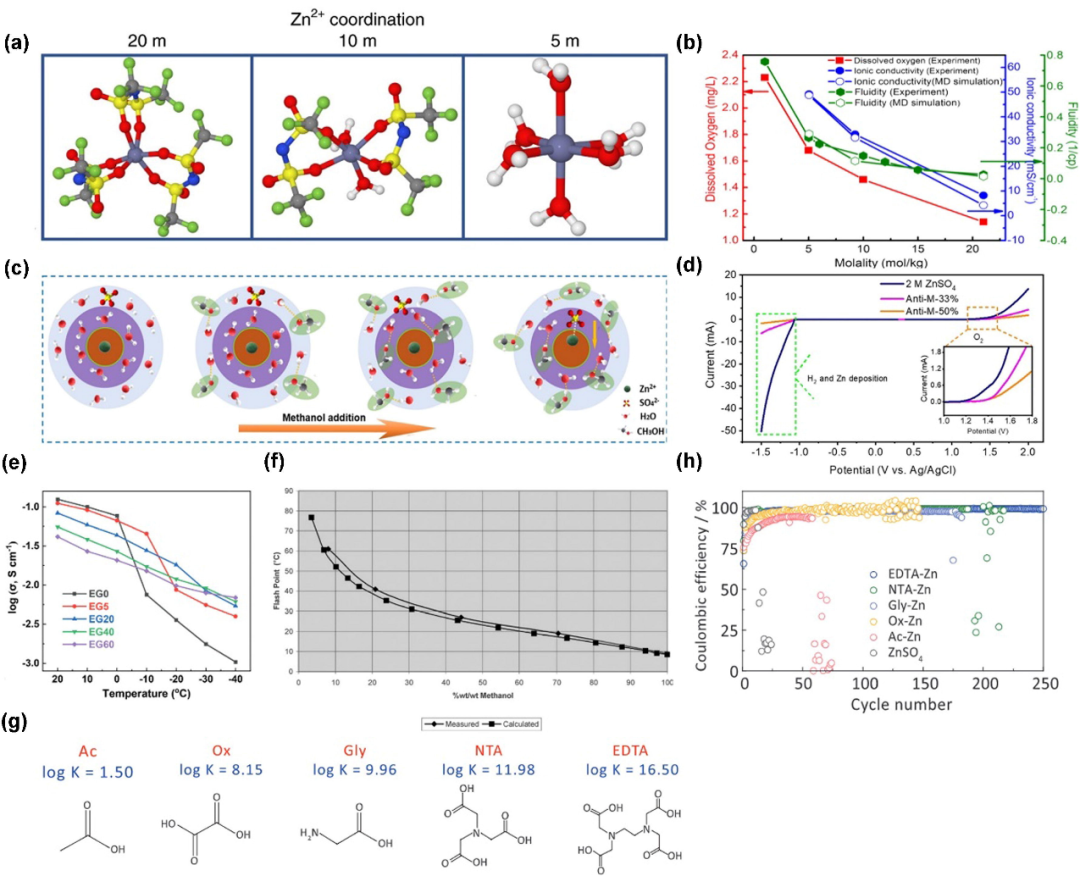
图3. (a)在1 m Zn(TFSI)2和3种LiTFSI浓度(5 m, 10 m和20 m)的电解质中Zn2+的溶剂化结构。(b)25°C时不同浓度电解质(LiTFSI-H2O)中Li+的电导率和流动性。(c)加入甲醇后Zn2+溶剂鞘的变化示意图。(d)不同电解质的LSV扫描,其中33%和50%代表甲醇的体积百分比。(e)不同EG/H2O比下电解质的离子电导率。(f)甲醇/水混合物闪点随浓度的变化。(g)不同K值配体的分子模型。(h)不同配体Cu/Zn半电池的CE稳定性。@Elsevier
电化学稳定窗口(ESW)
水较窄的稳定性窗口一直是水系电池的致命弱点,这也限制了AZIB的能量密度。为了扩大ESW,人们做了很多努力,如设计集流体、共溶剂和电解液净化。本节倾向于关注高浓盐水系电解质(WISEs)及其对体电解质电化学性能的影响。
电解质中的电荷载体被水溶剂化,无论是沉积在负极上还是嵌入正极,都在界面处脱溶剂。这种去溶剂化导致自由水在电场最强的界面处聚集,随后,水被电化学分解成H2(HER)或O2(OER)。通过引入高浓度,Zn2+溶剂化鞘中的自由水被阴离子取代(图3a),从而抑制了水的分解,最终扩大了ESW。谨慎的读者可能会在这里发现一个悖论,即Zn2+-阴离子对在前一节中损害了电解质的导电性,但在这里有利于ESW的扩展。事实证明,较高的浓度通常会导致许多WISEs的ESW扩展和电导率降低(图3b)。
按照同样的逻辑,研究人员可以实现类似的溶剂化鞘替换,而不必将浓度调得太高而不切实际。具有较高古特曼给体数的溶剂倾向于优先溶剂化阳离子,如将DMSO(给体数29.8)加入稀释的ZnSO4水溶液(H2O的给体数:18)中,用DMSO取代部分水,形成[Zn(H2O)4(OH)3(DMSO)]−等配合物。这样,电解质的阴极稳定性得到改善,库仑效率(CE)也随之提高。
各种有机溶剂被作为有效的电解质添加剂来扩大水系电解质的ESW。例如,添加50 v%的甲醇能够用于Zn2+溶剂化结构重建和拓宽ESW (图3c和3d),其他具有电子密集位点(通常为N或O基团)的有机溶剂也被广泛报道为助溶剂,如乙腈、乙二醇和1,4-二恶烷等。但需要指出的是,有机溶剂的加入也有其局限性。
例如,随着EG/H2O电解质混合物中EG含量的增加,离子电导率在室温下明显下降,这通常会导致更差的倍率性能和高电压滞后(有时被误解为更宽的ESW)。此外,添加有机溶剂带来的一个更致命的问题是电解质的可燃性更高,如图3f所示,当甲醇/水混合物中的甲醇浓度增加时,闪点低于室温。除助溶剂策略外,螯合剂可以在不改变水系理化性质的情况下与Zn2+产生较强的配位作用。Yang等人比较了各种螯合配体的稳定常数(K)作为Zn2+与配体结合强度的指标,结果表明,K值最高的乙二胺四乙酸通过更少的HER和最长的循环稳定性表现出更好的阴极稳定性(图3g和3 h)。然而,由于与Zn2+的螯合强度通常太强,Zn2+在溶液中无法保持电离,因此此类螯合剂添加剂应谨慎处理。
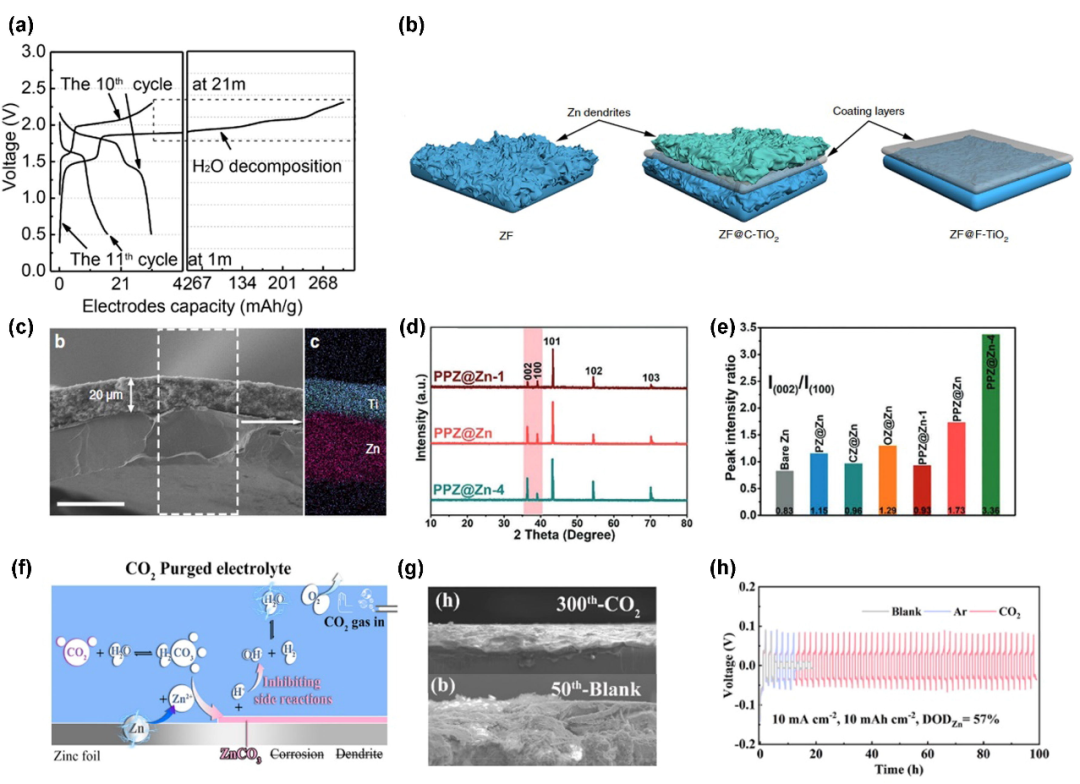
图4. (a)在WISE(21 m)中预形成SEI后,组装水系LiMn2O4/Mo6S8全电池。(b)不同TiO2晶面上的锌电镀过程示意图。(c)10次循环后(001)面涂层的SEM和相应的EDX映射图。(d)经磷酸处理1、2、4 min的Zn箔XRD谱图。(e)裸锌和锌compound@Zn样品的I(002)/I(100)比。(f)2 M ZnSO4电解液中Zn负极上的界面反应以及CO2净化电解液中这些副反应的抑制示意图。(g)CO2净化电解液(上)和空白电解液(下)中循环锌负极的SEM横截面图。(h)高放电深度下薄锌箔对称电池试验。@Elsevier
固体电解质界面相(SEI)
固体电解质间相(SEI)曾被认为是非水系电解质的特有产物,因为水的分解产物(H2和OH-)不会在负极上形成固体保护层。然而,在水系电解质中仍然观察到SEI,并且大多数与高浓度的WISE有关:首先,WISEs中阳离子和阴离子的接触离子对提高了阴离子的还原电位,这使得阴离子的分解优先于HER,构成了SEI的成分;其次,稳定的SEI需要最大限度地减少溶解。Wang等人的研究表明,即使是在20 m LiTFSI中稳定且结构良好的SEI,也会迅速溶解在稀释的1 m LiTFSI溶液中,导致不可逆性很高(图4a)。
除了浓度诱导的SEI外,人工SEI在保护锌负极方面也显示出一定的功能。对于一个好的人工涂层,主要有两个挑战:1、在温和酸性水环境下保持结构;2、允许Zn2+渗透沉积在涂层下,而不是生长在涂层上,同时阻止H2O的还原。Wang等人比较了TiO2各晶面Zn2+亲和力的差异。通过设计低Zn2+亲和力(001)晶面,进一步避免了Zn在涂层上的成核,确保了TiO2层下光滑均匀的Zn沉积形貌(图4b和c)。此外,磷酸锌与Zn的(002)面具有良好的晶格匹配度(图4d和e),这是一个众所周知的无枝晶外延生长晶面。因此,H3PO4诱导的(002)取向Zn生长延长了循环寿命,且形貌更加光滑。尽管证明了这种方法的有效性,但这种单相无机材料在沉积/剥离过程中Zn体积发生较大变化后,其刚性可能破坏结构的完整性。
电解质中溶解的CO2通常促进SEI的形成(图4f)。与LIB体系类似,用CO2清洗电解液可以获得更好的循环稳定性,形成富含Li2CO3的稳定SEI。Zhong等人报道了AZIB中类似的行为,与空白电解质的粗糙锌形貌相比,在CO2净化电解质中循环300次后,锌表面形成了致密的保护层。得益于这种涂层保护,即使在高放电深度(DOD)下,循环稳定性也大大提高,如图4h所示。
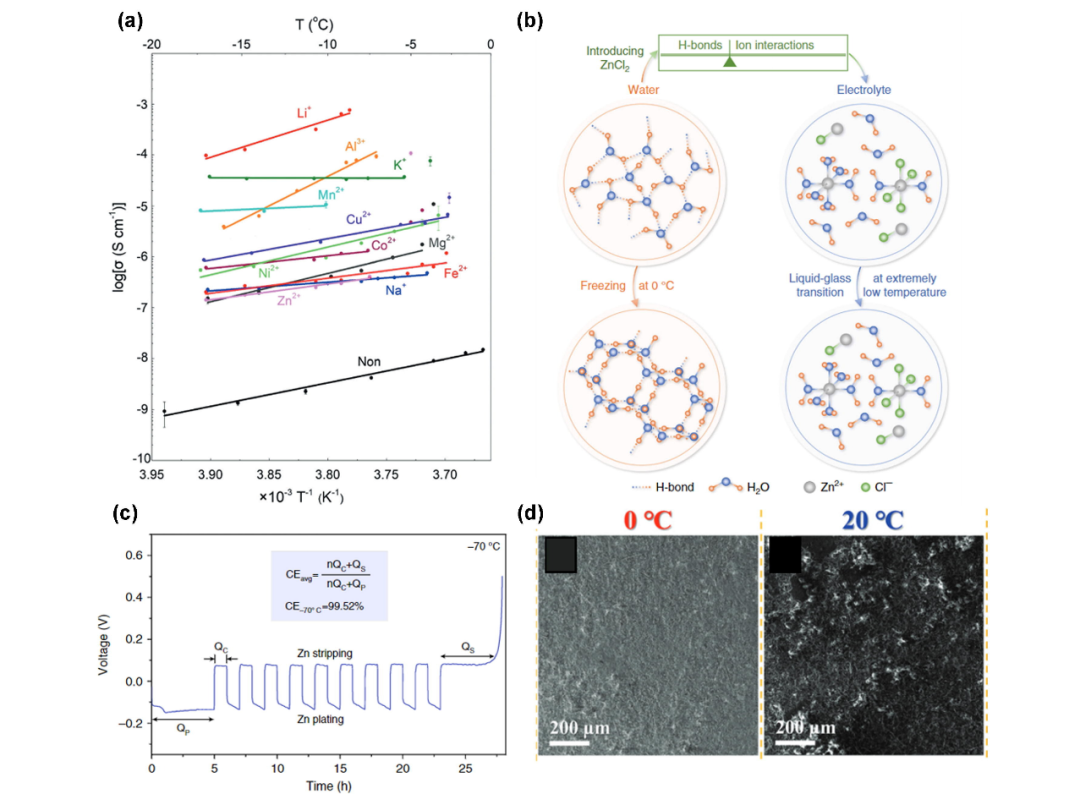
图5. (a)不同离子在冰中的Arrhenius离子电导率图。(b)水和电解质的结构演变示意图,以及低Tf溶液的设计。(c)非对称Zn/Cu电池在70℃时电镀/剥离锌的电压分布图。(d)Zn金属负极在0°C(左)和20°C(右)循环的SEM图。@Elsevier
低温
越来越极端的天气引起了人们对低温电池的关注。低温带来的电解质粘度增加等不利因素导致倍率性能变差,电解质冻结甚至电池失效。对于水系电池,尽管含有某些盐的冷冻电解质(冰)显示出高的离子电导率(Li2SO4为1 mS/cm),可能被用作固体电解质(图5a),但这仅仅是理论推测。一个更被广泛接受的策略是通过调整电解质浓度来调节电解质的冰点(Tf)。冷冻可以看作是水分子有序重组的结晶过程。通过添加电解质盐,H2O的O2-与Zn2+相互作用,而不是与其他水分子的H+相互作用,从而破坏分子间的氢键。减少的氢键网络为H2O的结晶创造了能垒,从而降低了Tf(图5b)。进一步增加浓度,形成阴阳离子对,使O2–-Zn2+相互作用减弱,Tf再次升高。按照这个思路,Chen等人通过溶解7.5 m的ZnCl2,将水系电解质的液态保持到-90°C。阳离子和阴离子在破坏H2O分子间氢键(HB)网络中都起着重要作用。有趣的是,由于抑制了HER反应,锌的沉积/剥离在低温下表现出更高的可逆性,并且沉积的锌形貌更光滑。7.5 m ZnCl2溶液的库仑效率从25℃时的97.93%提高到−70℃时的99.52%(图5c)。在3 M Zn(CF3SO3)2中进行了更多的形貌差异研究,其中循环Zn在0°C下保持光滑致密的表面,没有枝晶。而在20°C下循环的Zn表面变得粗糙和松散(图5d)。
04
成果启示
本综述从五个指标评估了AZIB电解质的作用,并揭示了不同电解质成分/浓度下电化学性能的巨大差异。从实用的角度来看,没有哪一种电解质配方能适用于所有的电池。较高浓度的电解质无疑会扩大ESW并促进SEI的稳定性,然而,它也会产生离子对,从而增加电解质粘度,降低离子电导率,提高溶液的冰点。对于高倍率性能和快速充电,Zn2+的快速传导优先于其他指标,并尽可能减小离子对/H+共传导。因此,超高浓度可能不适合高倍率性能。在低温方面,电解液浓度过低或过高都会导致水过早结晶,不能保持液态,阻碍Zn2+的传导。由于1、SEI形成带来更高的可逆性,而盐包水电解质导致水的反应性降低。2、在有限的电解液中加入更多的Zn2+可以补偿电极长期运行的不可逆性,所以WISE有望表现出更好的电化学性能。
审核编辑:刘清
-
固体电解质的物理性质如何?2019-09-17 2632
-
电池内的电解质是什么?2009-10-20 1234
-
锂离子电池聚合物电解质导电机理是什么?2009-10-29 7776
-
锂离子电池及其电解质的研究2009-11-04 3808
-
锂离子电池聚合物电解质导电机理2009-12-09 2811
-
电池电解液和电解质的区别_电池电解液和电解质的两种形态2020-04-16 25722
-
将商业化锂离子电池中的液态电解质替换什么解质?2020-06-09 3451
-
新型固体材料可替代电池中的易燃液体电解质2020-09-25 1426
-
锂离子电池堆电解质的要求及对电池性能的影响2020-12-30 5754
-
钠离子电池的电解质分类2022-10-09 6704
-
新型水系电解质实现长循环寿命的高压水系锂/钠离子电池2022-12-09 4399
-
弱溶剂间相互作用提高电池电解质稳定性2023-03-13 3488
-
分子筛电解质膜助力超长寿命锌离子电池2023-12-21 1905
-
不同类型的电池的电解质都是什么?2024-02-27 4226
-
锂离子电池电解质填充工艺:技术原理与创新实践2025-08-11 1503
全部0条评论

快来发表一下你的评论吧 !
