

高熵电解液赋能高可逆长循环镁金属电池
描述
背景介绍
可充电镁金属电池由于镁的高丰度、高容量和良好的安全性而受到了极大的关注。在其电化学还原/氧化过程中,Mg经历两次电子转移,可实现3833mAhmL-1的体积容量;Mg不容易形成Mg枝晶,这使得Mg金属负极比其他金属负极(如Li、Na和K)更安全。然而,表面钝化和缓慢的电化学动力学显著限制了镁金属负极的应用。镁金属负极表面形成的界面层通常是Mg2+绝缘的。形成活性[Mg2Cl3]+二聚体并同时避免表面钝化层的可行方法是将Cl引入电解质中。然而含Cl电解质的电化学窗口很窄,与商业集流体不兼容。因此,开发用于镁金属电池的无Cl电解质是一项重要的任务。
最近,引入了高熵概念来调节电解质的物理和化学性质,从而提高金属负极的电化学性能。引入多个组分以增加构型熵对自由能的贡献是抑制熵降低行为的有效方法,例如从混合溶液中分离一个组分和聚集两个或更多组分,大大提高了金属电极的电化学性能。
成果简介
近日,北京大学徐东升、李琦团队与中科大焦淑红团队受高熵电解质潜在优势的启发,开发了一种多组分高熵电解质,含有(Mg(TFSI)2、(DME)、(LiOTf)、TMP(磷酸三甲酯)),以稳定镁金属负极。由于OTf−和TMP的强配位能力,在Mg(TFSI)2/DME基电解质中获得了Mg2+-2DME-OTf--Li+-DME-TMP的高熵溶剂化结构。这种结构显著减少了Mg2+-DME的相互作用,促进了Mg2+导电层在镁金属负极上的形成,防止了Mg金属负极上绝缘成分的形成。该实验中,镁-铜电池表现出高达98%的高库仑效率,镁-镁对称电池在450小时内表现出低电压滞后。此外,Mg||Mo6S8全电池在200次循环内表现出稳定的电化学性能。这项工作首次设计并充分表征了一种能够成功稳定镁金属负极的高熵电解质。
图文导读 01 镁金属负极的电化学可逆性
DME中添加0.2MMg(TFSI)2被选为基础电解质。通过在基础电解质中加入LiOTf和TMP获得两种溶剂获得具有两种阳离子(Li+、Mg2+)、两种阴离子(TFSI-、OTf-)以及两种溶剂(DME、TMP)的高熵电解质。将Mg‖Cu电池与商用聚烯烃隔膜组装,以评估Mg金属负极的电化学可逆性。在最初的几个循环中,Mg金属负极在基础电解质中显示出近乎完全的不可逆性。当向基础电解质中添加10%(v/v)TMP时,CE在第一次循环中显著提高到58%,在40次循环后达到90%(图1a)。有趣的是,高熵电解质中的镁金属负极在随后的实验中表现出高达98%的CE。
图1b显示了Mg||Mg对称电池在0.1mA cm-2下在不同电解质中的电压-时间曲线。采用基础电解质的对称电池表现出约2V的大过电位,表明负极与电解质不相容;单独添加TMP可以有效地降低镁金属负极的电压滞后,但未能延长其使用寿命;在LiOTf和TMP的共同添加下,电池在最初的几个循环中表现出低得多的过电势,仅为80mV,450小时后过电势逐渐增加到580mV。因此,高熵电解质赋予电池的电化学性能,特别是其库仑效率、寿命、过电位和氧化稳定性,与从Cl或B基电解质中获得的结果相当。
图1c给出了不同电解质中镁金属负极的CV曲线,这一结果与对称电池的结果一致。在TMP和LiOTf的共同添加后观察到的强电流响应证明了这些添加剂对Mg金属负极的稳定性的协同作用。在1至0V的电压范围内获得的放大CV曲线揭示了镁金属成核之前电解质的电化学行为(图1d)。在TMP和LiOTf的共同添加后,出现了明显的还原峰,表明在Mg沉积之前形成了界面层。进行了电化学阻抗谱(EIS)来研究Mg金属负极的界面性质(图1e)。结果有力地证明了阳极上存在界面层。在高熵电解质中形成的界面层的电阻为996Ω,证实了其Mg2+导电性。

【图1】(a)Mg||Cu电池在Mg(TFSI)2/DME+TMP(有和无LiOTf)中的库仑效率。所施加的电流密度为0.5mAcm-2,并且面积容量为0.1mAh cm-2。(b)0.1 mA cm-2下,不同电解质中Mg||Mg对称电池的电压-时间分布。(c)Mg||Cu电池在三种电解质中以10mVs-1的扫描速率的循环伏安图。(d)Mg||Cu电池在电压范围为1至0V的三种电解质中的放大CV曲线
02 镁沉积的形貌和表面化学
为了揭示添加和不添加LiOTf/TMP的Mg(TFSI)2/DME的电化学性能的差异,作者使用透射电子显微镜(TEM)和扫描电子显微镜(SEM)研究了负极上形成的Mg沉积物的结构和形态。可以看出,在Mg(TFSI)2/DME中形成了多孔界面层(420nm)(图2a)。随着TMP的加入,界面层变得致密,其厚度降至220nm(图2b)。TMP和LiOTf共同加入到基电解质中导致了光滑紧凑的界面层,其厚度为140nm(图2c),利用聚焦离子束(FIB)-SEM对镁合金负极的形貌和结构进行了研究,在0.1mAcm-2的低电流密度和2h的长沉积时间下获得了大的镁合金镀层,并对其进行了表征(图2d和2e)。
与采用基础电解质的沉积物形成对比的是,添加LiOTf和TMP的Mg沉积物紧密堆叠,表面层薄(图2h、2i)。在Mg(TFSI)2/DME中形成了尺寸约为5μm的半球形Mg沉积物(图2f)。能量色散谱(EDS)图谱显示,大量的Mg和O分布在Mg沉积物上,这与TEM和FIB-SEM图像中观察到的厚界面层一致(图2g)。相比之下,在高熵电解质中形成的Mg沉积物光滑致密,具有明显的晶体取向(图2j,2k)。EDS图谱显示,大多数Mg沉积物和少量Au证实了其表面存在薄界面层。这种薄而紧凑的界面层具有高Mg2+电导率,有助于镁金属负极的高可逆性和长寿命。

【图2】Mg沉积的TEM图像,在(a)Mg(TFSI)2/DME、(b)Mg(TFSI)2/DME+TMP、(c)Mg(TFSI)2/DME+TMP/LiOTf;Mg在Mg(TFSI)2/DME上沉积的(d)FIB-SEM图像,(e)相应的放大SEM图像,以及(f)侧视图以及(g)顶视图;Mg在Mg(TFSI)2/DME+TMP/LiOTf上沉积的(h)FIB-SEM图像,(i)相应的放大SEM图像,以及(j)侧视图以及(k)顶视图;
采用X射线光电子能谱(XPS)和氩离子刻蚀技术研究了不同电解质下镁电极界面层的化学组成,组装了在0.5mAcm-2下10次循环后循环的镁-铜电池。首先,作者分析了高熵电解质形成的界面层的成分。N和S是阴离子的典型元素,可以反映TFSI或OTf−在DME和TMP中的还原程度。在Ar离子蚀刻前后,N1s信号可忽略不计(图3a)。考虑到TFSI是N元素的唯一来源,因此添加LiOTf和TMP限制了TFSI的降解。因此,-SOx(x≤2)的副产物必须来源于OTf−(图3b)。F1s光谱中685.4eV处的峰(图3c)、P2p光谱中134.3eV处的峰值(图3d)与Mg2p光谱中的重叠峰(50.84eV)一致,证明Mg3(PO4)2、MgF2和MgO在Mg表面上共存。近7%的可用P参与界面层,并且大多数P以Mg3(PO4)2的形式存在,这表明TMP在Mg电镀过程中被广泛还原,从而对界面层有贡献。总之,在高熵电解质中形成的外界面层由SOx和MgF2(源自OTf−)、磷酸盐(源自TMP)、MgO和有机醇盐组成。
在2分钟的Ar离子蚀刻后,Mg沉积物上的大量表面离子被去除。即使在2分钟的Ar离子蚀刻以去除表面离子之后,金属Mg仍然是Mg2p光谱中的次要离子。这一结果证实了在没有添加剂的情况下形成的界面层比在有添加剂的条件下形成的层厚得多。此外,TFSI阴离子在基础电解质中被严重还原,正如在N1s光谱所示(图3e)以及在S2p光谱所示(图3f)。即使在Ar离子蚀刻之后,这些峰的强度仍然保持,表明TFSI-的存在。与在高熵电解质中形成的界面层相比,在不含添加剂的基础电解质中形成的界面层更厚,并且含有大量TFSI衍生的物质(MgS、Mg3N2、N-SOx和SOx)和有机物(被DME还原),这会显著阻断Mg2+的扩散
元素N、S和F只能来自OTf−或TFSI阴离子;因此,作者总结了它们在所形成的界面层中的总比例,以评估阴离子的还原程度。在含有LiOTf和TMP的电解质中,Mg沉积物表面的N、S和F的总比例为7.25%,随后在Ar离子蚀刻后降至2.64%(图3h);基础电解质界面层中N、S和F的总比例为28.9%,几乎是高熵电解质中的四倍,Ar离子蚀刻后,该比例仍高达25.6%,表明TFSI在基础电解质中显著减少。TFSI的显著减少与Mg镀覆过程中的电化学条件高度相关。高电压滞后(~−2 Vvs.Mg2+/Mg)导致非常强的负电场,这将促进基础电解质中离解的TFSI的降解。相反,添加TMP和LiOTf后形成的优化界面层显著降低了电压滞后,缓解了TFSI-的降低。
利用TOF-SIMS来表征在高熵电解质中形成的界面层。PO4和Mg-P-O的存在有力地证明了Mg3(PO)4是在界面层中形成的(图3i,j)。利用傅立叶变换红外光谱(FT-IR)研究了Mg表面界面层的表面化学。如图3k所示,结果表明添加LiOTf和TMP前后,表面层的光谱显示出显著差异。TFSI和DME的信号明显减弱,Mg3(PO)4(P-O)出现强信号。这一发现证实了添加LiOTf和TMP前后界面层的显著差异。得益于添加LiOTf和TMP后形成的薄但致密的界面层,富含Mg3(PO)4的层表现出具有快速界面动力学的Mg2+导电性。相反,由于TFSI和DME的显著降解,基础电解质中形成的界面层非常厚,含有大量的MgF2、MgS、Mg3N2、MgO和有机物;这些物质显著阻断了Mg2+的扩散,并导致缓慢的电化学动力学。
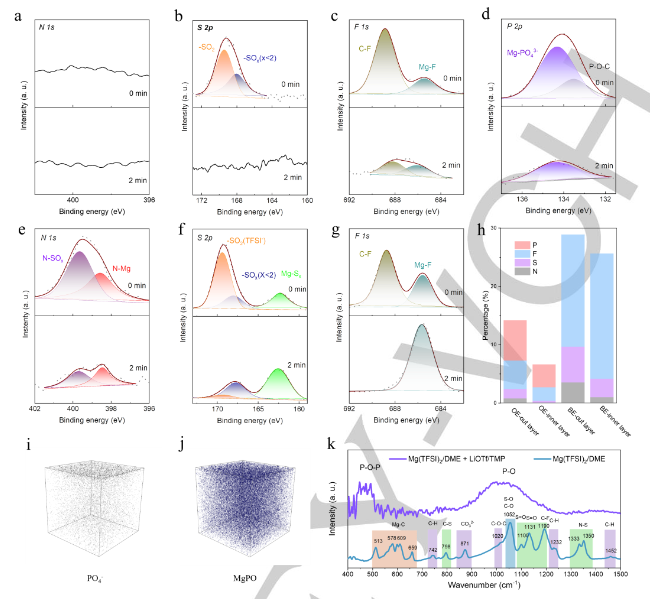
【图3】Mg(TFSI)2/DME与TMP和LiOTf(优化电解质,OE)形成的界面层的(a)N 1s、(b)S 2p、(c)F 1s和(d)P 2p XPS图谱。纯Mg(TFSI)2/DME(基础电解质,BE)中形成的界面层的(e)N1s、(f)S 2p和(g)F 1s XPS图谱。(h)Ar离子蚀刻前后两种电解质中界面层的原子比。TOF-SIMS深度剖面图和3D渲染图揭示了(i)PO4和(j)MgPO在Mg(TFSI)2/DME+TMP/LiOTf中沉积的Mg表面上的分布。(k)在含有和不含有LiOTf/TMP的Mg(TFSI)2/DME中形成的界面层的FT-IR光谱
溶剂化结构
界面层与电解质的溶剂化结构密切相关。因此,作者确定了两种电解质成分的供体数量,以预测它们的配位状态(图4a)。与TFSI-相比,DME分子更容易与纯Mg(TFSI)2/DME中的Mg2+配位。相反,OTf和TMP的供体数量相对较高,并且接近DME的供体数量。因此,这两种添加剂可以参与溶剂化鞘。核磁共振(NMR)结果表明,Li+-TMP和Mg2+-TMP的配位状态得到证实(图4b)。对这两种电解质进行了拉曼表征,可以观察到了配位TMP分子的存在。向电解质中添加LiOTf后,S=O(1286.8cm-1)的振动模式增加,证明了配位OTf的存在(图4c)。在Mg(TFSI)2/DME的拉曼光谱中,~881cm-1处的峰值对应于Mg(DME)32+,这与预测一致(图4d)。在添加TMP和LiOTf后,对应于Mg(DME)32+的峰的消失清楚地表明,由于添加剂的强配位能力,溶剂化鞘层发生了变化。
对Mg(TFSI)2/DME在LiOTf和TMP存在和不存在下的溶剂化结构进行了理论计算。纯Mg(TFSI)2/DME中的溶剂化鞘为Mg(DME)32+(图4e),这与拉曼光谱一致。值得注意的是,静电势的正电荷富集均匀分布在DME分子中的C-O键上(图4f),这意味着电子从DME中的O显著转移到Mg2+。这种富集将促进DME的还原并形成无活性的Mg(OCH3)2(DME)2物种。因此,Mg表面被大的过电位钝化,这进一步导致TFSI降解。然而,添加LiOTf和TMP后,溶剂化结构发生了显著变化。其中一个溶剂化的DME分子被OTf-取代,OTf-桥接Li+形成Mg2+-2DME-OTf--Li+-DME-TMP溶剂化结构(图4g)。因此,静电电势被重新分配。添加LiOTf和TMP后,DME的正电荷富集也发生了显著变化(图4h),最终削弱了从O到Mg2+的电子转移,缓解了DME的还原。此外,这种高熵溶剂化结构将OTf−和TMP带到了Mg电极的表面,并促进了它们的还原,这有利于构建Mg2+导电界面层。
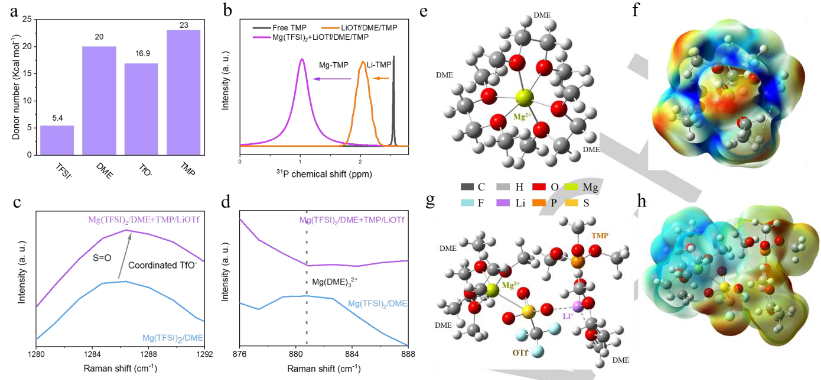
【图4】(a)TFSI-、DME、OTf-和TMP的供体数量。(b)纯TMP溶剂和TMP在不同环境中的31PNMR光谱。(c,d)两种电解质在(c)1280–1292 cm-1和(d)876–888 cm-1波数范围内的拉曼光谱。(e)典型的溶剂化鞘和(f)Mg(TFSI)2/DME中相应的静电电势分布。(g)典型的溶剂化鞘和(h)具有LiOTf/TMP的Mg(TFSI)2/DME中相应的静电电势分布。静电电势分布图中的红色和蓝色区域分别代表负电荷富集区和正电荷富集区
03 Mg||Mo6S8电池的电化学性能
将Mo6S8与镁金属负极组装,以评估高熵电解质的实用价值。如图5a所示,含有Mg(TFSI)2/DME的Mg||Mo6S8全电池在最初的几个循环后提供了约2mAhg-1的低放电容量。得益于高熵电解质,Mg||Mo6S8全电池表现出稳定的放电容量和超过200次循环的长寿命。含有Mg(TFSI)2/DME的Mg||Mo6S8全电池的库仑效率明显波动(图5b),而添加LiOTf和TMP的全电池的库伦效率保持相对稳定,并在200次循环中逐渐达到98.4%。这种效率比对照组的效率高得多。此外,电池的电压-容量曲线证实,充电/放电过程在高熵电解质中是稳定的(图S23a)。相比之下,在含有Mg(TFSI)2/DME的Mg||Mo6S8全电池中观察到约2mAhg-1的高过电位和低容量,这归因于所形成的绝缘界面层。尽管使用这种高熵电解质的Mg||Mo6S8全电池相对稳定,但这种全电池的衰变趋势归因于电解质的消耗和由此产生的电极极化。
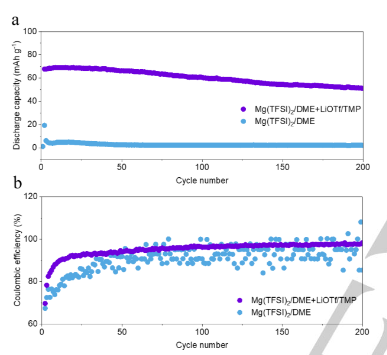
【图5】(a)Mg‖Mo6S8电池在两种电解质中的电化学性能和(b)库仑效率
总结与展望
通过在Mg(TFSI)2/DME电解质中共同添加LiOTf和TMP来稳定Mg金属负极。具有高供体数的OTf和TMP协同形成了高熵Mg2+-2DME-OTf-Li+-DME-TMP溶剂化结构,从而优化了去溶剂化过程,并在负极表面形成了富含Mg3(PO4)2的Mg2+导电膜。得益于高熵电解质,镁-铜电池提供了高达98%的高库仑效率。镁对称电池在初始循环期间提供了约80mV的低电压滞后,在450小时内逐渐增加到580mV。镁合金Mo6S8全电池的稳定电化学性能证明了高熵电解质设计在无氯镁金属电池中的潜在价值。
审核编辑:刘清
-
锂电池电解液如何影响电池质量?锂电池电解液成分优势是什么?2024-01-11 3089
-
电解液与SEI的关系?电解液对SEI的影响?2023-11-10 1941
-
稀释剂调节局部高浓电解液助力高电压锂金属电池2022-12-29 3673
-
解析高电压水系电解液最新研究进展2021-05-14 7290
-
锂电池电解液的组成部分_锂电池电解液的危害2020-08-03 12370
-
电池电解液和电解质的区别_电池电解液和电解质的两种形态2020-04-16 25745
-
锂电池电解液是什么_锂电池电解液主要成分2020-03-30 51007
-
电解液——锂电池的‘血液’2018-08-07 6116
-
锂电池电解液价格走势2018-03-08 12210
-
锂离子电池电解液超全面介绍 有何神秘之处?2017-02-22 7321
-
锂离子电池电解液是什么?2009-10-27 14819
-
高铁电池电解液2009-10-23 741
全部0条评论

快来发表一下你的评论吧 !
