

使用 RAPIDS 进行更快的单细胞分析
电子说
描述
单细胞测序已成为生物医学研究中最突出的技术之一。它在细胞水平上破译转录组和表观基因组变化的能力使研究人员获得了有价值的新见解。因此,单细胞实验的规模和复杂性增加了 100 多倍,涉及 100 多万个细胞的实验越来越普遍。
但是,必须在高度迭代的过程中对结果数据进行分析。至关重要的是,快速算法用于这些迭代步骤,以实现快速周转时间。
为了使用 Python 进行更一致的单细胞分析,scverse致力于构建一个完整的生态系统,以帮助研究人员进行分析。该生态系统的核心是一种数据结构,它可以在整个数据处理管道中维护各种转换的注释,从而实现单细胞分析。
AnnData 是一个 Python 包,用于处理内存和磁盘上的注释数据矩阵,是 Scanpy 库,是 scverse 生态系统中的主要单细胞分析套件。Scanpy 构建在 PyData 生态系统中常见的其他库之上,如 NumPy 、 SciPy 、 Numba 和 Scikit-learn,用于几乎所有典型的分析步骤。
然而, Scanpy 算法大多是基于 CPU 的,并且在较大的实验中速度显著减慢。单细胞分析过程的高度迭代性质只会加剧这个问题。
GPU 用于单细胞分析
RAPIDS 可用于 GPU 进行下游单细胞 RNA 测序(scRNA-seq)分析的一般可行性,这一可行性已在 《使用 GPU 加速单细胞基因组分析》 中得到证实。此外,该工作还产生了 rapids-single-cell-examples GitHub repo,其中包含一系列由 RAPIDS 和 NVIDIA Parabricks 构建的示例。RAPIDS 是一个 GPU 的开源库套件,用于 Python 的加速数据科学,而 Parabricks 是一套免费的 GPU 加速的、基于深度学习的行业标准基因组分析。
虽然这些示例笔记本在 GPU 上展示了一些典型的单细胞 RNA 工作流程,但它们从未用于日常使用,也从未被用作像 Scanpy 这样的库的 GPU – 加速替代品。
从以前的工作中汲取灵感,一个新兴的图书馆叫 rapids-singlecell,是一种用于 scRNA 分析的 GPU 加速工具。该工具旨在成为与 scverse 生态系统兼容的每日可驱动单细胞分析套件,它使用 RAPIDS 和 CuPy 以提供 GPU 加速的函数,这些函数几乎是 Scanpy 中相应函数的替代品。更多信息可以参考 rapids-single-cell-examples。
一般来说,用户可以期望通过使用 RAPIDS,将性能提高 10 到 20 倍。想要了解更多信息,请访问 使用 RAPIDS 加速单细胞基因组分析。
使用 RAPIDS 进行更快的单细胞分析
RAPIDS-singlecell 遵循与 scverse Python 库类似的可用性模型。它也是用 Python 编写的,但将许多性能关键部分放在 GPU 上,隐藏了通常与编写 CUDA 应用程序(通常用于为 NVIDIA GPU 编写加速算法的语言)相关的所有复杂性。
RAPIDS-singlecell 由五类组成,将在以下章节中进行描述。每个类别都加速了典型的单细胞分析工作流的不同部分。
想了解更多信息,包括 RAPIDS-singlecell 提供的各种 API,请访问 rapids-singlecell 文档。
cunData
AnData ,或注释数据对象,是一种广泛使用的数据结构,用于处理单细胞 RNA 测序数据。相反, cunData 是 GPU 的 AnData 对象的最小化和轻量级版本,它取代了用于预处理的 scverse 标准(图 1 )。 cunData 不是将计数矩阵. X 存储在 CPU 上,而是将其作为 CuPy 稀疏矩阵存储在 GPU 上。这使得对计数矩阵执行计算更快、更高效。它还包含细胞(. obs 属性)和基因(. var 属性)的注释数据帧,用于存储细胞类型和基因名称等附加信息。
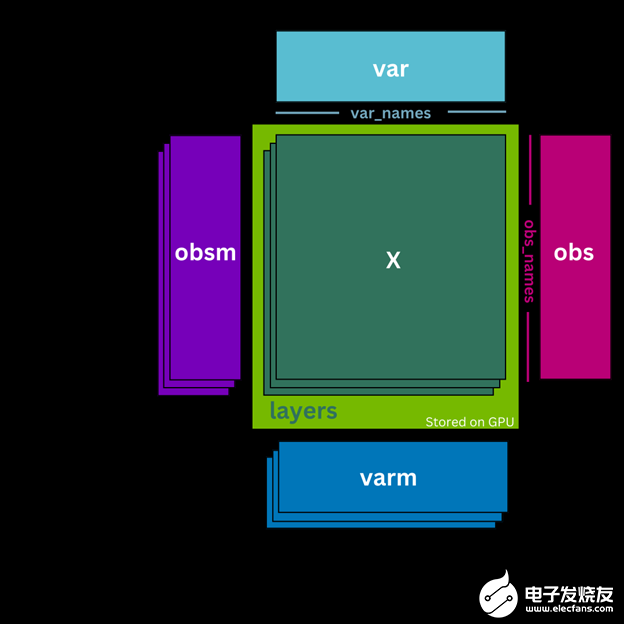 图 1 。 cunData 类结构示意图
图 1 。 cunData 类结构示意图
cunData 还包括其他功能,例如能够将不同版本的计数矩阵(如原始整数计数)存储在. Layers 中。与将. Layers 存储在主机( CPU )内存中的 AnData 不同, cunData 还在 GPU 上存储. Layers ,从而减少了将数据从主机复制到 GPU 内存的需要,并实现了加速计算。
cunData 支持. uns 属性中的非结构化注释,以及. obsm 和. vrm 属性中的细胞和基因的多维注释,这些注释存储在主机内存中。这些注释使用户能够包括关于其数据的附加信息,例如空间坐标或主成分分析( PCA )嵌入。
类似地, cunData 支持像 AnnData 一样的切片。但是,与视图相反,这些切片始终是原始数据的完整副本。总体而言,与更具特征丰富的 CPU 结合的 AnnData 对象相比, cunData 能够更快地预处理 scRNA-seq 数据。
下面的 Python 片段演示了 AnnData 对象(用于处理单细胞 RNA 测序数据的标准数据结构)到 cunData 对象的转换。
import scanpy as sc
import rapids_singlecell as rsc
adata = sc.read("PATH TO DATASET")
cudata = rsc.cunnData.cunnData(adata=adata)
预处理
预处理函数存储在cunnData_funcs,为 Scanpy 预处理功能提供了加速的替代方案。这些函数在cunData对象,并使用 RAPIDS cuML 和 CuPy 来显著加速基于 Scikit-learn 、 NumPy 和 SciPy 的 Scanpy 函数。
过滤细胞和基因可以用filter_cells和filter_genes功能。质量控制由calculate_qc_metrics作用
# Basic QC rapids-singlecell rsc.pp.flag_gene_family(cudata,gene_family_name="MT", gene_family_prefix="mt-") rsc.pp.calculate_qc_metrics(cudata,qc_vars=["MT"]) cudata = cudata[cudata.obs["n_genes_by_counts"] > 500] cudata = cudata[cudata.obs["pct_counts_MT"] < 20] rsc.pp.filter_genes(cudata,min_count=3)
为了规范化您的数据, cunData _ funcs 提供了 GPU 替代normalize_total, log1p,以及最近推出的normalize_pearson_residualsScanpy 的函数。注释highly variable genes对于 Scanpy 支持的所有口味(包括seurat,cellranger,seurat_v3,pearson_residuals)以及poisson_gene_selection,它改编自 scvi 工具。
# log normalization and highly variable gene selection cudata.layers["counts"] = cudata.X.copy() rsc.pp.normalize_total(cudata,target_sum=1e4) rsc.pp.log1p(cudata) rsc.pp.highly_variable_genes(cudata,n_top_genes=5000,flavor="seurat_v3",layer = "counts") cudata = cudata[:,cudata.var["highly_variable"]==True]
这个regress_out用 cuML 线性回归估计器加速了用于去除不必要的变异源的函数。它还支持多目标回归,这是在版本 22 . 12 的 cuML 中引入的,同时与以前的版本保持向后兼容。
cuML 包装的主成分分析(PCA)、截断奇异值分解(Truncated SVD)和增量主成分分析(Incremental PCA)为您提供与 Scanpy 提供的 PCA 函数 相同的选项。在 cunnData_funcs 中的 PCA 版本中,您可以选择要用于分析的图层,这是 Scanpy 目前不支持的附加功能。
# Regression, scaling and PCA rsc.pp.regress_out(cudata,keys=["total_counts", "pct_counts_MT"]) rsc.pp.scale(cudata,max_value=10) rsc.pp.pca(cudata, n_comps = 100) sc.pl.pca_variance_ratio(cudata, log=True,n_pcs=100)
cunndata_funcs可以将预处理加速 10 到 20 倍(表 1-3 )。经过预处理后, cunData 对象被转换为 AnnData 对象。
adata_preprocessed = cudata.to_AnnData()
工具
scanpy_gpu提供了在 AnData 对象上工作的函数,目的是提供加速的函数。在 Scanpy 和 RAPIDS -singlecell 之间保持尽可能接近的语法,元数据也被写入.uns属性该属性对于存储经过训练的参数(例如在 PCA 计算期间计算的方差比)非常有用。scanpy_gpu为 AnnData 对象提供了一个 PCA 函数,相当于cunnData_funcs.
Scanpy 已经支持使用 cuML 计算 GPU 上的 UMAP 和最近邻居。scanpy_gpu通过添加更多算法,如基于加速图的算法,扩展了对 Scanpy GPU 的支持clustering使用Leiden和Louvain来自 cuGraph ,以及Force Atlas 2用于直观地布置图形数据的算法。scanpy_gpu也使用PCA和kernel density estimation( KDE )来自 cuML 和diffusion maps以类似于 Scanpy 使用 SciPy 和 NumPy 进行科学计算的方式,使用 CuPy 库进行计算。
对于批量校正,scanpy_gpu提供的 GPU 端口Harmony Integration,可以调用harmony_gpu。PyMDE(最小失真嵌入),一种能够嵌入单细胞数据,同时以概率方式联合学习图和低维表示的函数,也可以在scvi-tools中使用。
RAPIDS – 单细胞置换性质的接近下降可以使用 Scanpy 进行可视化,使用起来很直观,Scanpy 用于绘图可以直接在 scverse 框架内进行。
解耦器
这个decoupler工具使用统一的框架实现了几种不同的统计方法,重点关注生物活动(如细胞、分子和生理过程,例如基因集和转录因子活性)。decoupler_gpu重新实现并加速了加权和 (run_wsum) 以及多元线性模型 (run_mlm) 方法。RAPIDS 中的 GPU 端口-singlecell 使用与解耦器相同的网络/模型。表 1 显示 wsum 的性能提高了 37 倍。
Squidpy 开发
RAPIDS – 单细胞正在不断扩展,为 scverse 生态系统提供新的加速功能。库中添加了全面的测试,以确保代码的正确性和可靠性。 Squidpy 能够对空间分子数据进行详细分析和可视化。它有助于理解复杂的细胞相互作用和空间模式,极大地促进了 scverse 生态系统的扩展。
RAPIDS 加速了一些功能,空间自相关与莫兰的 I 和 Geary 的 C 承诺性能提升高达 100 倍。配体受体 (ligrec) 在 Squidpy 中的交互分析也得到了优化和加速,性能提升超过 10 倍。
基准
我们的基准测试结果表明,将 GPU 加速与 RAPIDS – 单细胞包和解耦器功能一起使用,可以显著提高 scRNA-seq 分析的性能。
例如,运行示例 rapids-singlecell notebook,在具有两个 AMD Epyc Milan 7543500GB 内存和一个NVIDIA A100 80GB GPU 的环境下,使用 RAPIDS-singlecell 包仅需 51 秒即可完成,而传统的扫描 CPU 工作流程仅需 1106 秒。
类似地,解耦器功能也显示出显著的速度改进,与 CPU 上的 83 秒相比, GPU 上的 mlm 功能仅运行了 12 秒wsum方法在 GPU 上只需 26 秒,而在 CPU 上只需要 16 分 10 秒。
总之,这些结果证明了 GPU 加速使 scRNA-seq 分析更快、更有效的潜力。表 1 总结了这些基准结果。
| 作用 | CPU | GPU | 加速 |
| 整个笔记本电脑(不包括 PR 功能) | 1106 秒( 18 . 5 分钟) | 51 秒 | 21 倍 |
| 预处理 | 74 秒 | 8 秒 | 9 倍 |
| HVG (Seurat v3) | 27 秒 | 1 . 6 秒 | 16 倍 |
| Regress out | 35 秒 | 0 . 7 秒 | 50 倍 |
| scale | 3 . 2 秒 | 0 . 4 秒 | 8 倍 |
| PCA | 417 秒 | 18 秒 | 23 倍 |
| Neighbors | 22 秒 | 5 . 1 秒 | 4 . 3 倍 |
| UMAP | 36 秒 | 0 . 4 秒 | 90 倍 |
| TSNE | 133 秒 | 2 . 4 秒 | 55 倍 |
| Louvain | 17 秒 | 0 . 6 秒 | 28 倍 |
| Leiden | 14 秒 | 0 . 2 秒 | 70 倍 |
| 逻辑回归 | 58 秒 | 3 . 7 秒 | 15 倍 |
| 绘图( FA2 ) | 256 秒 | 0 . 3 秒 | 850 倍 |
| run_mlm ( DoRothEA ) | 83 秒 | 12 秒 | 7 倍 |
| Run_wsum (程序) | 970 秒( 16 分钟) | 26 秒 | 37 倍 |
表 1 。 90000 个单元格数据集的服务器节点基准测试
除了之前的基准测试结果之外,运行一个示例rapids-singlecell 笔记本,当使用 RAPIDS-singlecell 时,服务器节点上的 500K 个 cell 大约只需要 2 分钟。而在 CPU 上进行同样的分析则需要 41 分钟。
此外,使用pearson_residuals对于高度可变的基因选择和标准化,也可以使用 GPU 加速,从而在 scRNA-seq 分析中提供额外的速度改进。这些基准结果汇总在表 2 中。
RAPIDS-singlecell 不仅能够在高端服务器节点上加速单小区数据分析,而且能够在消费级硬件上加速singlecell 数据分析。在 AMD 5950x CPU 、 64GB 内存和 NVIDIA RTX 3090 GPU 的台式机系统上,使用 RAPIDS-singlecell ,端到端运行具有 50000 个电池的同一笔记本电脑大约需要 5 分钟。尽管系统使用RAPIDS Memory Manager (RMM)和统一内存来超额订阅 GPU 内存,但与 CPU 服务器相比,它的速度仍然显著提高。这些基准结果汇总在表 2 中。
| 作用 | CPU | GPU ( A100 ) | GPU ( 3090 ) | 加速 |
| 整个笔记本(不包括公关功能) | 2460 秒( 41 分钟) | 110 秒 | 290 秒 | 22 倍 |
| 预处理 | 305 秒 | 28 秒 | 169 秒 | 10 倍 |
| HVG (Seurat v3) | 48 秒 | 1 . 5 秒 | 13 秒 | 32 倍 |
| Regress out | 104 秒 | 5 . 1 秒 | 16 秒 | 20 倍 |
| scale | 8 . 4 秒 | 1 . 3 秒 | 5 秒 | 6 . 4 倍 |
| PCA | 86 秒 | 3 . 7 秒 | 35 秒 | 23 倍 |
| Neighbors | 74 秒 | 17 . 1 秒 | 18 . 3 秒 | 4 . 3 倍 |
| UMAP | 281 秒( 4 . 6 分钟) | 6 . 7 秒 | 7 . 6 秒 | 60 倍 |
| TSNE | 786 秒( 13 分钟) | 10 秒 | 12 . 9 秒 | 105 倍 |
| Louvain | 283 秒( 4 . 5 分钟) | 4 . 5 秒 | 5 . 7 秒 | 62 倍 |
| Leiden | 282 秒( 4 . 5 分钟) | 0 . 6 秒 | 0 . 9 秒 | 470 倍 |
| 逻辑回归 | 452 秒( 7 . 5 分钟) | 33 秒 | 63 秒 | 13 倍 |
| 扩散贴图 | 30 秒 | 0 . 75 秒 | 1 . 3 秒 | 40 倍 |
| 重型车辆(公共车辆) | 104 秒 | 2 . 1 秒 | 15 . 6 秒 | 50 倍 |
| 规格化( PR ) | 22 秒 | 0 . 3 秒 | 1 秒 | 73 倍 |
表 2 。 500000 个单元格数据集的服务器节点和消费者系统基准
当使用 RAPIDS -singlecell 时,在桌面系统上端到端运行具有约 90K 个单元格的相同示例笔记本(表 1 )仅需 48 秒。相比之下,传统的扫描 CPU 工作流程需要 774 秒。加速解耦器功能还显示出在消费级硬件上的显著速度改进。表 3 总结了这些基准结果。
| 作用 | CPU | GPU | 加速 |
| 整个笔记本电脑(不包括解耦器功能) | 774 秒( 13 分钟) | 48 秒 | 16 倍 |
| 预处理 | 114 秒 | 6 秒 | 19 倍 |
| Regress out | 62 秒 | 1 . 6 秒 | 39 倍 |
| 主成分分析 | 42 秒 | 0 . 7 秒 | 60 倍 |
| HVG (Seurat v3) | 2 . 7 秒 | 0 . 4 秒 | 6 . 7 倍 |
| PCA | 175 秒 | 21 . 7 秒 | 8 倍 |
| Neighbors | 14 . 9 秒 | 4 . 6 秒 | 3 . 2 倍 |
| UMAP | 31 秒 | 0 . 3 秒 | 103 倍 |
| TSNE | 95 秒 | 1 . 4 秒 | 68 倍 |
| Louvain | 9 . 3 秒 | 0 . 5 秒 | 18 倍 |
| Leiden | 13 . 2 秒 | 0 . 1 秒 | 130 倍 |
| 逻辑回归 | 76 秒 | 3 . 75 秒 | 20 倍 |
| 绘图( FA2 ) | 191 秒 | 0 . 23 秒 | 830 倍 |
| run_mlm ( DoRothEA ) | 55 秒 | 12 秒 | 4 . 5 倍 |
| Run_wsum (程序) | 690 秒( 11 . 5 分钟) | 28 秒 | 26 倍 |
表 3 。 90000 个细胞数据集的消费者系统基准
安装
安装 RAPIDS – 单电池有多种方法。最简单的方法是使用 GitHub 存储库中提供的一个 yaml 文件。这些设置了整个环境,包括运行示例笔记本所需的一切。
conda create -f conda/rsc_rapids_23.02.yml
您还可以从 PyPI 将 RAPIDS -singlecell 安装到 Conda 环境中,并从 Conda 安装 RAPIDS 。默认安装程序不包括 RAPIDS 或 CuPy 。 Scanpy 也被排除在外,因为它在技术上是不必要的。
pip install rapids-singlecell
最后,您可以使用 RAPIDS 中的新实验 PyPI 包从 PyPI 安装整个库,包括 RAPDIS 依赖项。然而,这种安装方法需要用户正确设置 CUDA ,以便可以通过 RAPIDS 和 CuPy 找到它。
要执行此操作,可以使用以下命令:
pip install 'rapids-singlecell[rapids]’ --extra-index-url=https://pypi.nvidia.com
结论
带 RAPIDS-singlecell 库·,可以在比 CPU 仅计算其 UMAP 嵌入所花费的时间更短的时间内运行 500K 个小区的完整分析。因此,它能够在单细胞数据分析阶段实现更快的迭代过程。
RAPIDS-singlecell 还使生物信息学家能够与医生或生物学家实时分析数据,从而更好地协作和解释数据。根据我们的经验,即使在消费类 3090 系列显卡上,也可以在没有任何问题的情况下分析 200K 单元。更好的是, RMM 使 GPU 存储器能够被超额订阅并溢出到主存储器,从而使规模远远超过 500K 个单元。
使用数据中心类 NVIDIA A100 80 GB GPU ,您可以分析包含多达 2 个的矩阵31-1 (约 21 . 5 亿)个非零计数。(请注意,这是用于稀疏矩阵计算的基于 CuPy 32 位整数的索引的当前限制。)这种强大的功能使用户能够分析超过 100 万个单元格的数据集。
20 倍以上加速了 RAPIDS-singlecell 提供的功能使研究人员能够更加专注于分析和解释他们的单细胞数据,而不是等待漫长的计算过程。本着 RAPIDS 的真正精神,这最终提高了生产力,并促进了对细胞生物学的新见解,这在以前是不可能的。
-
单细胞分析仪中步进电机的定位控制_金路2017-01-17 903
-
微流控芯片在单细胞捕获中的应用2019-04-08 9918
-
什么是高通量单细胞RNA测序技术?2019-04-25 11468
-
微流控芯片单细胞克隆形成抑制实验用于乳腺癌干细胞特异性药物筛选2019-06-20 6755
-
厦门大学研发出全新高通量单细胞转录组测序方法2020-06-02 4776
-
单细胞转录组学技术的十年2020-09-23 4958
-
“解码”单细胞测序的故事2021-03-17 3257
-
使用RAPIDS在NVIDIA GPU上分析脑细胞基准2022-04-18 2272
-
通过RAPIDS加速单细胞DNA和RNA基因组分析2022-04-27 2174
-
基于微流控平台的单细胞通讯研究进展2022-11-29 1946
-
单细胞+转录组测序:揭示GABA信号调控神经发生2023-01-12 1960
-
CellTrek单细胞空间转录组联合分析2023-04-01 3706
-
可用于单细胞分析的选择性液滴提取微流控装置2023-04-10 2014
-
北京基因组所开发出新型高通量单细胞多组学技术2023-04-20 2275
-
一种用于微液滴中单细胞无标记分析的液滴筛选(LSDS)方法2024-03-26 1504
全部0条评论

快来发表一下你的评论吧 !
