

具有可逆阴、阳离子氧还的Na-Mg-Fe-Mn-O正极
描述
01
导读
钠离子电池(NIB)有望用于电网规模的储能应用。然而,含钴、镍的正极材料使得它们的成本效益较低。而层状正极材料由于其低成本和易于合成而成为多种材料的有吸引力的替代品。特别是,基于Fe和Mn的NaxTMO2由于Fe和Mn的丰富性和低成本而引起大家的关注。
02
成果简介
针对上述问题,来自中国科学院物理研究所胡勇胜&容晓晖&肖东东团队、美国麻省理工学院李巨团队将Mg2+成功地用于激活Fe/Mn基层状正极中的氧氧化还原反应,以实现可逆的混合阴离子和阳离子氧化还原能力。在没有O损失的情况下实现了≈210 mAh g−1的高首次充电容量和平衡的充放电效率,显示出2.02 美元 kWh−1的有前景的能源成本。没有预钠化的全电池对硬碳负极显示能量密度超过≈280 Wh kg−1,在100次循环后具有85.6%的良好容量保持率。还对电荷补偿机制和结构演变进行了全面分析。确认了部分可逆的Fe3+迁移到Na层导致的电压和容量损失,揭示了低成本NIB正极在应用场景中的进一步改进。相关工作以题为“Earth-Abundant Na-Mg-Fe-Mn-O Cathode with Reversible Hybrid Anionic and Cationic Redox”的论文在线发表在Advanced Energy Materials上。
03
关键创新
(1)晶体结构
通过简单的固相反应合成了O3型NaFe0.5Mn0.5O2和O3型Na0.83Mg0.33Fe0.17Mn0.50O2样品。NaMFM样品的晶体结构通过NPD和nPDF测试得到确认,这些测试也使用Rietveld方法进行了改进,如图1a、b所示。如图1c所示,场发射扫描电子显微镜(FESEM)显示NaMFM具有典型的层状形状,尺寸分布在2–4 μm范围内。此外,X射线能量色散谱(EDS)映射(图1d)结果表明,Na、Mg、Fe、Mn和O在整个层状颗粒中均匀分布。
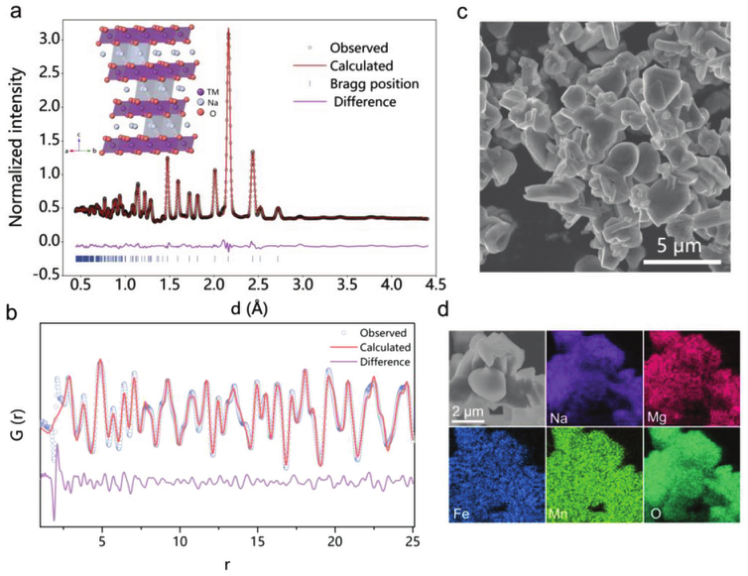
【图1】 O3-NaMFM的结构、形态特征。a)O3-NaMFM的精细NPD模式。插图:NaMFM晶体结构示意图。b)nPDF模式和Rietveld精修配置文件。c)NaMFM的SEM图像。d)Na、Mg、Fe、Mn和O元素的EDS映射。
(2)电化学性能
设计的NaMFM和NaFM作为钠离子电池正极材料的电化学性能针对钠金属负极进行了研究。图2a显示了NaMFM和NaFM在10mAg−1下的典型充电和放电曲线,NaMFM表现出一个斜率,伴随着归因于Fe3+/Fe4+和氧氧化还原的明确电压平台,与CV曲线一致,从而证明了轴向Na-O-Mg构型提供的阴离子-氧化还原活化,提高了那些特定O 2p轨道的轨道能量。NaMFM在不同电压范围内的循环性能如图2b所示。对于NaMFM,放电容量随着截止电压的增加而增加。多次相变和Fe迁移可能是导致NaFM循环性能差的原因。NaMFM作为正极材料的可行性在没有任何预钠化的情况下针对硬碳负极在全电池中得到进一步证明(图2c)。全电池表现出更好的循环性能,在100次循环后容量保持率为85.6%(图2d)。这是由于在全电池系统中HC上形成了相对稳定的SEI层,而不是半电池中Na负极上的SEI层反复形成和分解。
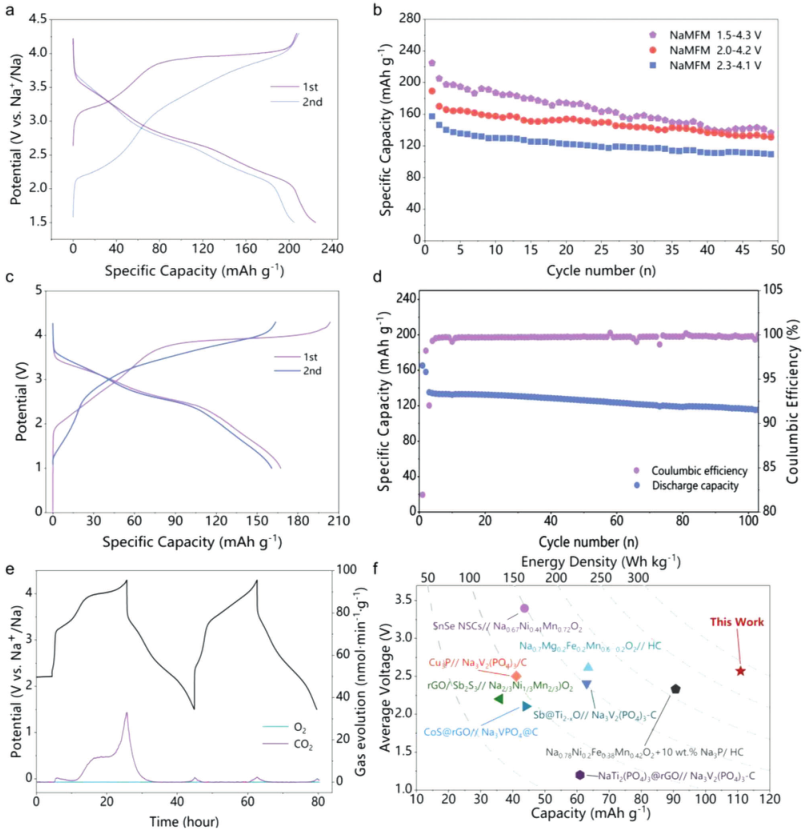
【图2】O3-NaMFM的电化学性能。a)在10 mA g−1下,Na/NMFM在1.5和4.3 V之间的恒电流充电和放电曲线。b)10 mA g−1下,不同循环窗口之间恒电流循环性能的比较。c)在10 mA g−1下,NaMFM/硬碳全电池的恒电流充放电曲线。d)NaMFM//硬碳全电池在50 mA g−1下的循环性能,全电池在10 mA g−1下预循环。e)NaMFM在电化学(脱)钠化过程中的气体损失。在10 mA g−1的前两个NaMFM循环期间收集的原位DEMS数据。上图显示电池的电化学响应,下图显示随时间变化的O2和CO2气体。f)NaMFM//硬碳全电池与其他已报道的钠离子电池全电池系统的能量密度比较,这些系统是根据正极和负极中活性材料的总质量得出的。
为了进一步检查氧气是否从晶格中流失,在前两个循环期间进行了差示电化学质谱法(DEMS)。DEMS测试的结果显示在图2e中,在4.3 V充电时未检测到O2气体,因为仅观察到CO2,与之前的研究相对应。总之,无法检测到不可逆的氧损失,表明NaMFM晶格中没有O损失,Mg2+离子在实现可逆氧氧化还原反应中起着稳定作用。
(3)电荷补偿机制
为了了解Fe、Mn和O在不同电化学过程中所起的作用,通过多种非原位光谱技术测试了具有不同电化学状态的NaMFM样品。在Fe和Mn的K边收集非原位hXAS光谱以研究价态变化和局部结构演变,并应用在Fe、Mn L2,3边和O K边的非原位sXAS来确定TM 3d和O 2p的电子结构在各种Na(脱)嵌状态下的变化,并了解电荷补偿机制(图3b、c)。结果表明,Fe离子的氧化主要负责第一次充电期间的电荷补偿。然而,在高达4.3 V的初始充电过程中,氧阴离子的氧化还原反应提供了额外的容量。
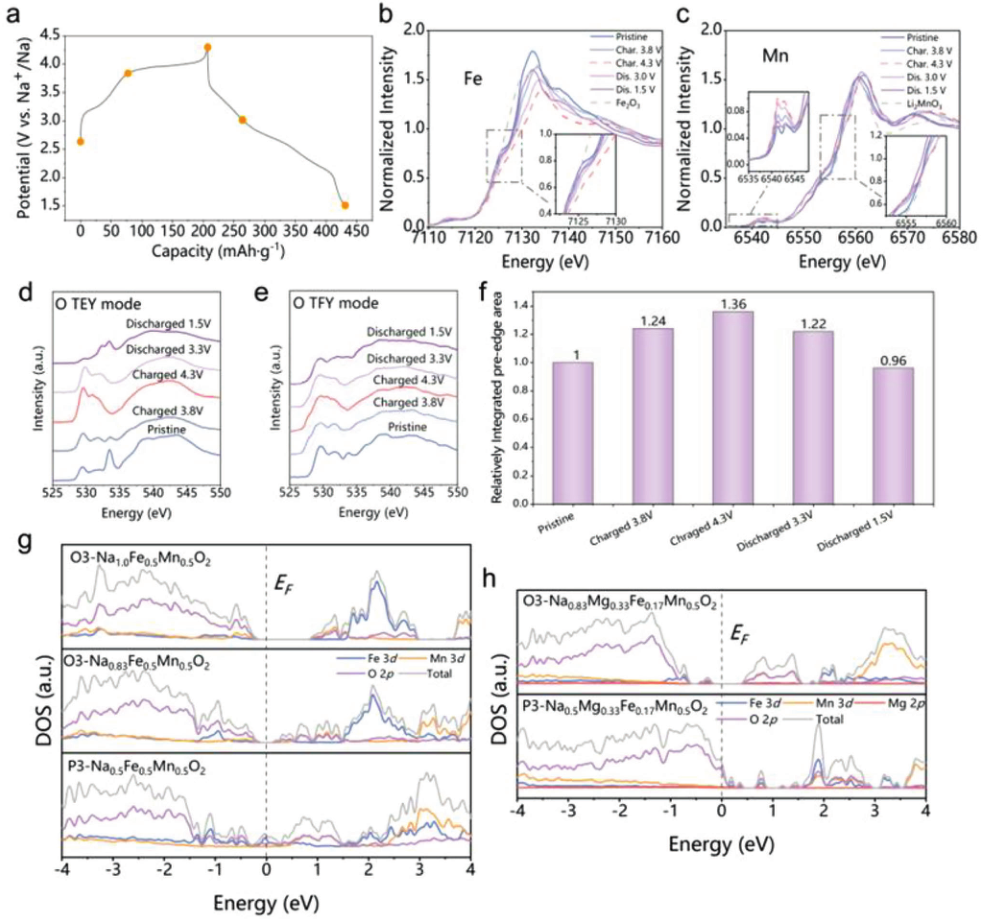
【图3】循环过程中NaMFM的电荷补偿机制。a)用于非原位XAS测试的NaMFM电极的充放电曲线和对应点。b)Fe和c)NaMFM在第一个循环中处于不同状态的Mn K边XANES。d)TEY模式和e)TFY模式的O K边软XAS光谱在不同NaMFM状态下。f)在从TFY模式导出的第一个循环期间,O K边XAS的前边(从528到533 eV)下的积分区域的比较。g,h)NaFe0.5Mn0.5O2和Na0.83Mg0.33Fe0.17Mn0.5O2的原始样品,以及Na0.83Fe0.5Mn0.5O2、Na0.5Fe0.5Mn0.5O2和Na0.5Mg0.33Fe0.17Mn0.5O2的带电样品的预计态密度(PDOS)。
为了研究氧氧化还原可能参与电荷补偿机制,测试了O K边的sXAS的总荧光产率(TFY)模式和总电子产率(TEY)模式(图3d、e)。在图3f中,可以看出充电过程中光谱下方区域的前边峰发生变化,表明O 2p空穴和平均有效电荷显著增加,这意味着氧气有助于充电过程中的电荷补偿。在放电过程中,前边峰的积分面积减小,表明MFM中氧氧化还原的高可逆性。正如O K边XAS光谱所证明的那样,在充电和放电期间的整个电压范围内,MFM中都存在可逆的氧氧化还原。
为了进一步了解Mg掺杂在氧化还原机制中的作用,进行了密度泛函理论(DFT)计算,以获得NaFe0.5Mn0.5O2和Na0.83Mg0.33Fe0.17Mn0.5O2原始样品以及Na0.83Fe0.5Mn0.5O2、Na0.5Fe0.5Mn0.5O2和Na0.5Mg0.33Fe0.17Mn0.5O2带电样品的预计态密度(PDOS),如图3g、h所示。Na0.5Mg0.33Fe0.17Mn0.5O2中费米能级附近O p轨道贡献的电子态密度远大于Na0.5Fe0.5Mn0.5O2中的电子态密度,表明O2−/O−的阴离子氧化还原活性更高。此外,通过比较Na0.5Fe0.5Mn0.5O2和Na0.5Mg0.33Fe0.17Mn0.5O2的PDOS,观察到Mg在提高费米能级附近O p轨道的电子密度方面起着主导作用,从而激活O2−/O−的阴离子氧化还原活性。
(4)晶体结构演化
为了阐明Na+嵌入/脱嵌引起的反应机理和NaMFM晶体结构的变化,在前两个循环中进行了原位XRD,结果如图4a所示。与原始O3相相关的(003)和(006)峰移动到较低的角度并分裂成两个,随后出现一组与较低角度的六方P3相相关的新峰。新峰的移动和出现表明,当Na+被提取时,由于相邻氧层之间的排斥力增加,c轴扩展,并且在同一区域,(104)O3峰移动到更高的角度,因为面内TM–TM键长减小,表明过渡金属被氧化。当电池持续充电至3.4 V时,O3峰完全消失,所有衍射线都归属于P3相,一直持续到电池充电至4.0 V,显示该区域为单相反应,与斜率变化曲线。随着Na+的进一步提取,观察到一个新相“X”,X相呈现宽峰,其强度随着Na的去除而降低。当进一步充电到最后时,≈17°处的峰值在脱层后连续移动到更高的角度,表明层间距离收缩。收缩可能与阳离子迁移和O的氧化相关,导致排斥力降低。如图4a所示,观察到几种不同的不对称衍射图案,特别是在X区域。这种不对称展宽是由TM层的系统滑动引起的,这可能导致堆垛层错。放电后,XRD经历了从“X”到O3的直接演化而没有转变为P3,并且与原始O3相的峰相比更宽的衍射峰意味着在一个完整的循环后结晶度降低,这可能与Fe迁移有关。
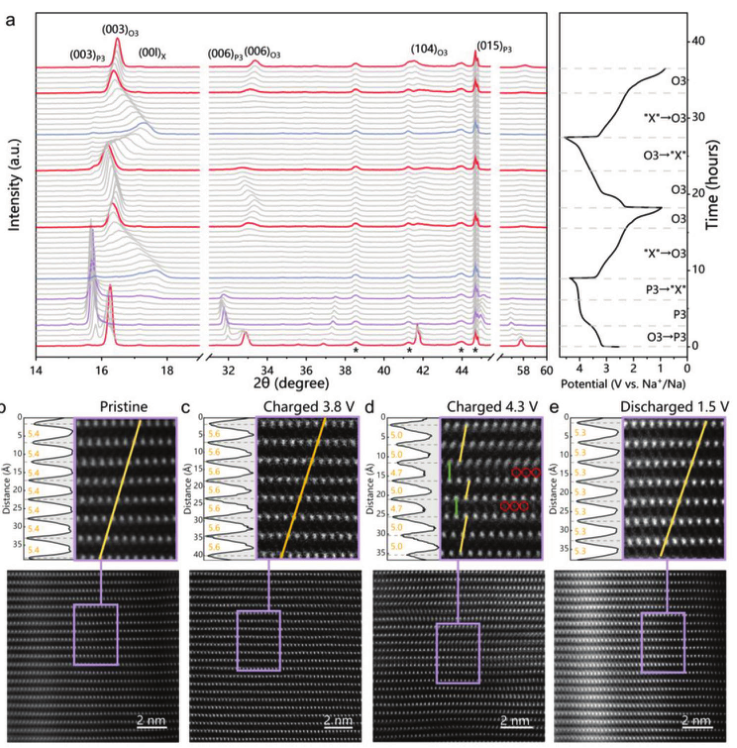
【图4】结构演变。a)在O3-NaMFM电极的前两次充电/放电过程中收集的原位XRD图案在1.5和4.3 V之间循环,电流为20 mA g−1。从原始NaMFM和三种不同的充电状态获得的原子分辨HAADF-STEM图像:b)原始,c)充电至3.8 V,d)充电至4.3 V,和e)放电至1.5 V。所有图像都是沿[100]投影。每个图像的突出显示区域清楚地显示了层的堆叠,这些层被识别为P型(无TM偏移:绿线)或O型(TM偏移:橙色线)。强度变化绘制在每个突出显示图像的左侧,以测试相邻TMO2层之间的层间距离。
进行了HAADF-STEM成像,以原子分辨率可视化“X”相。图4b–e说明了原始NaMFM和三个非原位样品(充电至3.8 V、4.3 V并放电至1.5 V)沿[100]投影的TMO2层的堆叠。图4c显示了充电至3.8 V的样品的HAADF-STEM图像。相比之下,充电至4.3 V的样品图像(图4d)显示对齐的相邻层(绿线)和偏移的相邻层(黄线),可以分别用O1和P3相索引。这意味着X相由P3和O1结构域组成。当放电至1.5 V(图4e)时,有序的O3堆叠序列将恢复,层间距约为5.3 Å。与原始样品相比,层间距离较小是由于放电过程中Mn4+的减少所致。
MS用于更清楚地了解Fe在相变和电荷补偿中的作用。对于NaMFM组合物,原始材料(图5a)、完全充电材料(图5b)和完全放电材料(图5c)的光谱是在室温下在密封条件下获得的。异构体位移(IS)、四极分裂(QS)和Fe3+/Fe4+离子的相对浓度如表1所示。结果表明,在第一次充电过程中发生的Fe迁移大部分是可逆的。结合在HAADF-STEM图像中观察到的阳离子迁移,这可以解释原位XRD中显示的不可逆相变,即保留在Na层中的残余Fe锚定了层间滑移并阻断了Na扩散路径,从而导致在前两个循环中观察到容量损失和电压滞后现象。
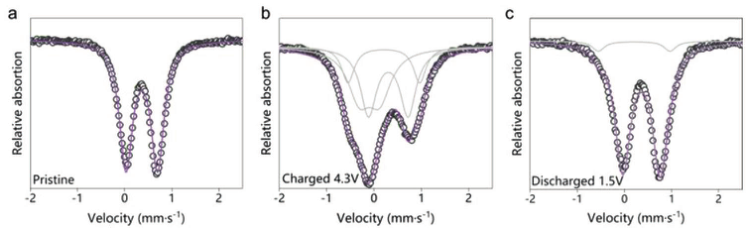
【图5】不同充电状态下的57Fe-Mössbauer光谱。a)原始状态,b)充电至4.3 V,c)放电至1.5 V。原始数据以黑色显示。来自拟合的Fe3+O6、Fe3+O4和Fe4+O6的各个光谱分量以灰色显示,紫色线表示总拟合。
【表1】57Fe-Mössbauer光谱参数。
05
成果启示
报告了一种潜在的不含镍和钴的铁基和锰基钠离子正极材料,无需额外的牺牲盐或电化学预钠化即可实现全电池运行。Fe和Mn基正极由于地球上丰富的Fe/Mn而特别具有吸引力,并且此类正极在资源方面与高度依赖Li、Ni和Co的LIB技术具有互补性。不完全可逆迁移Fe是这种材料中大电池极化和结构失效的主要原因,必须加以解决,以提高其未来应用的性能。即,可以通过掺杂元素增加Fe迁移的活化能或通过抑制在高压下发生的P-O相变来抑制它。这些发现为设计具有高结构稳定性的可逆氧氧化还原正极材料提供了新的机会。
审核编辑:刘清
-
具有优越循环性的双重改性的低应变富镍正极软包全电池2025-01-07 3190
-
课题组JACS:O3-NaNi1/3Fe1/3Mn1/3O2的结构演变解析2024-12-04 2723
-
卡拉胶作为5V高压LiNi0.5Mn1.5O4正极牺牲粘合剂!2023-08-01 2680
-
O2型富锂正极中阳离子超晶格有序性对阴离子氧化还原的影响2023-02-03 3054
-
揭示粘结剂对LNMO正极性能衰退的影响机理2022-09-28 3223
-
利用粘结剂PAN提高TM离子不可逆迁移的能量势垒2022-08-30 5867
-
钠离子电池层状正极材料结构稳定性和氧化还原电位的精准调控2022-08-02 7464
-
去离子器基本原理及应用概述2021-11-25 13706
-
超氧-过氧化物之间的可逆转化2021-01-14 3643
-
锂过量的正极材料中可逆的Mn2+/Mn4+双氧化还原2020-12-25 1618
-
钠离子电池正极材料怎样实现可逆氧变价的结构2019-02-10 2273
-
景区负氧离子监测系统,生态保护区环境负氧离子监测系统,森林公园负氧离子浓度监测设2018-08-08 1032
-
阳离子交换器作用及工作原理2017-10-26 19508
-
锂离子电池的最新正极材料:掺锰铌酸锂?2016-01-19 6407
全部0条评论

快来发表一下你的评论吧 !
