

一系列同构环状夹层化合物的设计、合成和表征,并将其命名为“环烯”
描述
01 研究背景
二茂铁的分离及其分子结构的解释标志着现代有机金属化学的起源。在这一里程碑式的发现之后,夹层配合物及其衍生物对化学本身的发展和理论基础产生了无与伦比的影响。带有两个环戊二烯配体(Cp,C5H5-)的夹层化合物通常被称为茂金属。不过,在当今的科学术语中,茂金属的严格定义并不是强制性的。特别是在 f 元素的情况下,具有较大配体系统的化合物,如环辛四烯二酰胺,也被称为茂金属。使用高维含金属网络或特殊形状结构的概念在有机金属化学中并不陌生。然而,尽管经过 70 多年的研究,夹层化合物(其中的结构完全由金属-配体直接相互作用组装而成)仍局限于简单的链或相关图案。除此之外,还有一些基于二茂铁或二茂钌衍生物的环状低聚金属聚合物。其中,通过 SiMe2-、CHR-(R = H、Me)或 C-C 键等配体-配体键提供的互连框架已被报道。然而,这些物质并不具有任何多层结构模式。
02 研究问题
本研究展示了一系列同构环状夹层化合物的设计、合成和表征,并将其命名为“环烯”。这些环烯由 18 个重复单元组成,在固态下形成几乎理想的圆形闭环,可用通式 [cyclo-MII(μ-η8:η8-CotTIPS)]18 描述(M = Sr、Sm、Eu;CotTIPS = 1,4-(iPr3Si)2C8H62-)。量子化学计算得出的结论是:离子金属与配体之间的键、配体系统的体积和闭环时的能量增益之间的独特相互作用促进了这些环状系统的形成。迄今为止,只有线性一维多十克夹层化合物被研究用于纳米线等可能的应用领域。这一环状夹层化合物的教科书范例有望为进一步创新新型功能性有机金属材料打开大门。
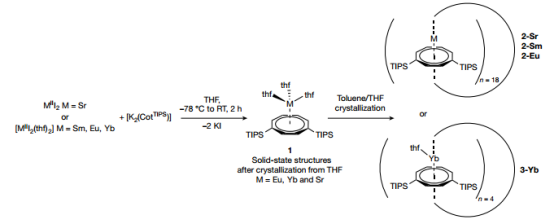
▲图1 | 环烯合成
要点:
1.二价碱土和镧系碘化物 SrIII2 或 [MIII2(thf)2](M = Sm、Eu、Yb)与 [K2(CotTIPS)]在四氢呋喃(THF)中的反应生成了单体 THF 溶剂 [MII(thf)3(η8-CotTIPS)]配合物(1,M = Sr、Eu、Yb)。所有络合物在移除母液时都会迅速失去配位的 THF 分子(图 1)。Sm络合物无法获得结晶物质。1 的固态分子结构类似于之前描述的相关复合物 [YbII(PYR)3(η8-Cot)](PYR,吡啶)的钢琴凳图案。由于 1-Sr 和 1-Eu 在干燥分离出的晶体材料时会迅速失去所有溶剂分子,因此对 1 进行全面表征的尝试并不成功。而 1-Yb 则保留了一个等量的 THF。本研究首次证明:在干燥 1-Sr 或 1-Eu 时会形成 [LnIIn(CotTIPS)n]的不含 THF 的化合物。因此,在结晶过程中必须抑制 THF 配位,以便形成只包含 MII-Cot 相互作用的低聚系统。然而,在使用甲苯或正庚烷等非配位溶剂的几次结晶尝试中,得到的都是无定形物质。在甲苯结晶批次中加入少量的四氢呋喃最终促进了单晶的分离。单晶 X 射线衍射分析表明,离子半径相近的金属离子 SrII、SMII 和 EuII 形成了环十八碳夹层配合物[cyclo-MII(μ-η8:η8-CotTIPS)](2,图 2)。
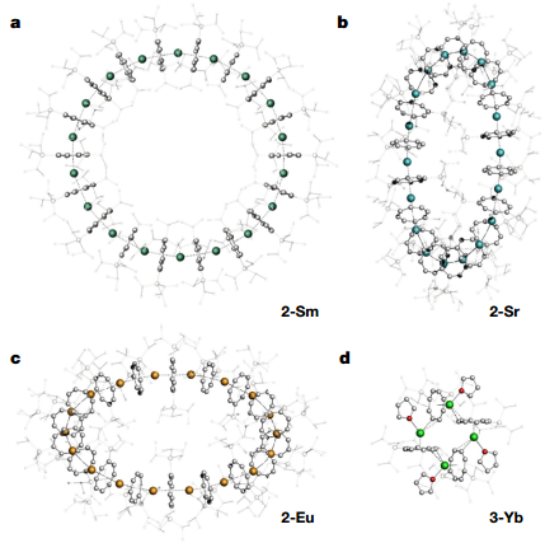
▲图2|环烯的结构
要点:
1.通过使用较小的 YbII 离子,本研究得到了正式的环四叔丁烯 [cyclo-YbII(μ-η2:η8-CotTIPS)(thf)]4(3-Yb,图 2,右下角),其中 YbII 配位球的开口面被一个 THF 配体占据。本研究认为,与 SrII、SMII 和 EuII 相比,κ1-THF-YbII 键得到了加强,这是因为 YbII 的路易斯酸度更高。因此,与环十八碳夹层结构相比,收缩结构更受青睐。
2.为了探究化合物 2 在溶液中的结构组成,本研究对抗磁性复合物 2-Sr 和顺磁性复合物 2-Eu 进行了扩散有序核磁共振光谱(DOSY-NMR)实验。在 THF-d8 中,这些实验得出的流体力学半径分别为 Rh = 6.9 Å(2-Sr)和 5.3 Å(2-Eu),表明存在[MII(thf)3(η8-CotTIPS)] (1) 类型的单体物质。在非配位溶剂甲苯-d8 中,流体力学半径为 Rh = 5.3 Å(2-Sr)和 5.0 Å(2-Eu)。尽管对不同溶剂中的 DOSY 测量值进行比较时应谨慎考虑,但这些值明显偏离了溶液中存在环十八碳夹层结构的预期值。因此,环状图案很可能是在结晶过程中通过自组装途径形成的。同样,二茂铅[Pb(Cp)2]根据结晶条件的不同,可以形成不同的链或带有 Pb-η5-Cp-Pb 桥的六聚环状支架。不过,铅原子由三个 η5-Cp 环配位,因此并不像经典的夹层图案。
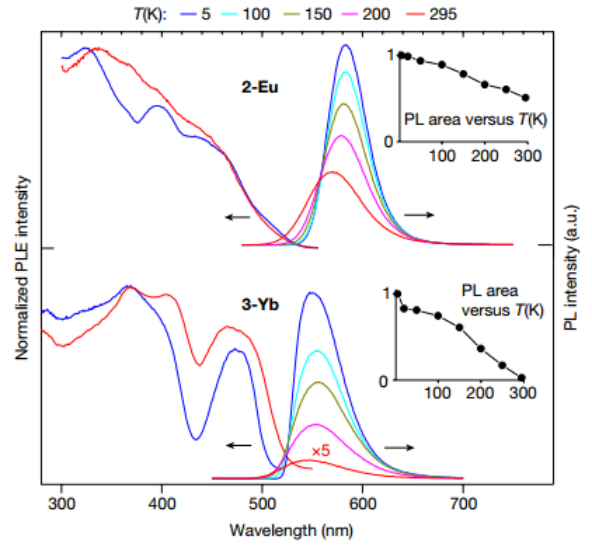
▲图3|光致发光特性
要点:
1.固体环烯 2-Eu 和 3-Yb 发出的橙色和绿色光致发光(PL)的中心波长分别为 570 纳米和 546 纳米,在低温条件下,光致发光的中心波长略微偏移至 583 纳米和 550 纳米(图 3)。激发起始波长约为 520 nm,这与样品的黄色(2-Eu)或黄绿色(3-Yb)多晶粉末的外观以及吸收光谱一致。在室温下,2-Eu 的发射量子效率高达 ΦPL = 60%(估计在 5 K 时接近 100%),在 5/295 K 时,337 nm 的 ns级脉冲激光激发下,2-Eu 的发射量子效率呈单指数衰减,τ = 2.1/1.7 µs。因此,它只显示出适度的温度依赖性。在二价Eu的其他复合物(包括夹层化合物)中也观察到了类似的 PL,可归因于 2-Eu 中 Eu2+ 离子的 4f65d1 → 4f7 转变。低能吸收带或 PLE 带也与 Eu2+ 的 f-d 转变有关,因为预计 CotTIPS 配体的分子轨道在大约 400 nm 以上的吸收波长或 PL 激发波长不会产生影响。相关化合物中二价Eu和镱的发射 f-d 转变的能量通常相当。从光谱上看,2-Eu 和 3-Yb 的发射非常接近(图 3)。
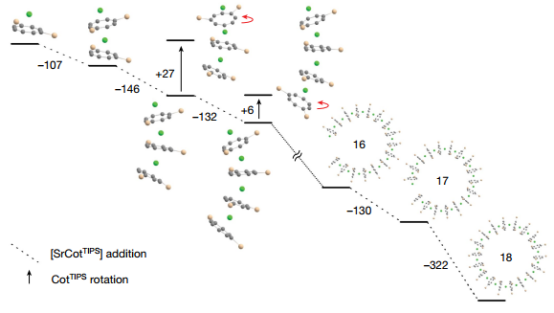
▲图4|量子化学计算
要点:
1.显然,取代基的立体需求是造成单个环-M-环单元弯曲的原因。然而,这种立体诱导的弯曲只有在单向发生时才会形成所观察到的环状结构。否则,也可能形成链状结构。通过考虑图 4 中部分描述的 Sr 模型系统,本研究关注了这两种选择之间的差异。计算得出以下结果:
(1) 与不适合形成环的最低能量排列相比,代表环分子部分的排列在能量上具有 6 至 27 kJ mol-1 的增量。
(2) 根据 n = 2 到 n = 17 时 [Sr(CotTIPS)]n-1 + [Sr(CotTIPS)] → [Sr(CotTIPS)]n 的反应能量估算,形成环的能量增益约为 140 kJ mol-1。对于环闭合的宽(n = 18),最低能量是原来的两倍多,为 322 kJ mol-1。
(3) 对于适合成环的排列方式,分散相互作用只比不适合成环的排列方式略大(2-16 kJ mol-1)。然而,分散相互作用占反应能量的一半以上,因此其对稳定结构至关重要。
03 结语
环烯家族[cyclo-MII(μ-η8:η8-CotTIPS)]18(M = Sr、Sm、Eu)的分离和结构特征研究揭开了夹层化合物化学的新篇章。在量子化学方法的帮助下,这些迷人化合物的形成过程被合理化,并转化为基本的设计原则,为今后合成环烯奠定了基础。在环烯结构中观察到的明显弯曲是由 Cot 取代基的立体需求造成的。环形成的驱动力是其闭合所获得的能量,这在很大程度上受弥散相互作用的影响,而当形成非环结构图案时,这种能量就会消失。环烯的稳定性和大小可以通过特别定制所使用的配体系统来调节。考虑到经典夹层化合物的这一前景及其公认的特性,这种环烯图案的教科书范例有望在功能有机金属化学中得到进一步应用。
-
华林科纳的化合物半导体异质集成2023-08-14 1511
-
有机化合物可作为锂离子电池正极材料2015-11-17 5623
-
labview如何把一个数组(或者矩阵)命名为A2018-01-15 3233
-
浅析化合物半导体技术2019-06-13 6159
-
金属间化合物观察与测量2020-02-25 1287
-
III-V族化合物,III-V族化合物是什么意思2010-03-04 4474
-
媒体确认新款iPad被命名为iPad HD2012-03-06 1068
-
新一代iPhone命名为iPhone8?错了,是iPhoneX!2017-02-06 3886
-
华为下一代智能手表或命名为WatchX2018-09-19 5482
-
从英国化合物半导体中心看化合物半导体集群2019-04-11 6582
-
三星下一代Galaxy S系列或命名为Galaxy S20系列2019-12-31 3499
-
iPhone 13系列或将命名为iPhone 12s2021-01-15 2389
-
传苹果2021年新款手机或命名为iPhone 12s2021-01-19 3151
-
英伟达新一代芯片或将命名为“CMP”2021-02-19 2726
-
无金属参与的可见光催化醛和铵盐合成腈类化合物2023-09-27 2391
全部0条评论

快来发表一下你的评论吧 !
