

三合一策略实现废旧LiCoO2正极高效再生!
描述
研究背景
作为一种能量密度高、使用寿命长、成本低、自放电能力低的具有高度代表性的前沿技术,锂离子电池(LIBs)广泛应用于便携式电子设备、电动汽车和网格级储能系统。在所有LIBs正极材料中,LiCoO2(LCO)由于其高压平台、能量密度高、大规模生产方便等固有优势,在便携式电子设备中占主导地位,占电池总重量的30%。据统计,世界各地每年废弃的笔记本电子设备生产超过10万吨,如果处理不当,将造成严重的环境危害,造成宝贵金属资源的巨大浪费。更糟糕的是,由于资源短缺,原材料价格的上涨给电池生产商和材料供应链带来了太大的压力。实际上,废弃的LIBs可以作为许多金属元素的次要来源。因此,迫切需要将更多的注意力转移到废旧钴酸锂(D-LCO)的可持续能源存储上。现有的热法冶金和湿法冶金工艺一般涉及多步骤的净化和分离过程,缺乏经济可行性和环境友好性。此外,上述两种方法都是破坏性的回收过程,即正极材料内的能量被完全破坏。因此,探索一种绿色、节能、无损的直接再生D-LCO材料的途径仍需要不懈的努力。
成果简介
近日,合肥物质科学研究院张云霞/湖州师范学院韩苗苗针对D-LCO的有效再生提出一个直接的无损策略,通过固相烧结的方法,对D-LCO 的成分和结构缺陷进行修复,通过对D-LCO进行表面改性以及Mn、N和S元素的掺杂,解决D-LCO组成/结构缺陷、氧释放和结构扭曲等内在问题,优化其循环性能,保证了再生后的LCO类似或优于原始材料的电化学性能。在此,作者通过通过各种表征技术,系统地研究了降解和循环的LCO材料的组成和晶体结构的演化。除了Li补偿外,Mn和N/S元素分别成功地掺杂到Co位点和Li-O结构中,同时也实现了Li2SO4在LCO粒子表面的原位涂层。对再生后的LCO的速率能力和循环稳定性进行了评价,并与商业LCO和D-LCO进行了比较,在高比容量、速率能力和良好的循环稳定性方面有显著提高。此外,还应用原位X射线衍射(XRD)、Rietveld细化分析和第一性原理计算(DFT)来探讨充放电过程中的固有结构演化和潜在的再生/修饰机制。该工作以“Upcycling ofDegraded LiCoO2 Cathodes into High-Performance Lithium-Ion Batteriesvia a Three-In-One Strategy”为题发表在Advanced Energy Materials上。
研究亮点
(1) 设计一种非破坏性的一锅工艺,使D-LCO直接转化为高能量密度的MNS-LCO正极材料,具有易于放大的潜力;
(2) 基于所开发的三合一固相烧结策略,可以有效地恢复D-LCO的组成和晶体结构,同时实现Li2SO4原位表面涂层和Mn掺杂Co位,N、S掺杂Li-O板。;
(3) 改性策略能有效减少O2逃逸,抑制有害相变,稳定重建LCO的层状结构.
图文导读
基于先前报道的失效分析,D-LCO正极材料的能力衰减主要归因于缺乏锂离子和结构崩溃造成的长期循环,导致氧逃逸,寄生与电解质反应和石盐和尖晶石相等结构缺陷。本文提出了一种无损再生技术,通过Li补偿和分层结构修复,对D-LCO材料进行直接升级。上循环过程如图方案1所示。简单地说,从退役计算机上提取的D-LCO袋电池首先进行彻底的放电处理(图S1,支持信息),然后进行物理拆卸。然后,D-LCO正极材料很容易从铝箔中剥离。随后,将收获的D-LCO活性粉末直接分散在含(CH3COO)2Mn和硫脲的乙醇溶液中,并不断搅拌。在溶剂完全蒸发后,收集的样品进一步与适量的Li2CO3机械混合,在300°C下退火半小时,然后增加到850°C,在空气中进行10小时,以达到所需的结晶度并抑制结构缺陷。值得注意的是,在热退火过程中,由于Mn、N、S元素的加入,锂缺陷LCO粒子的缓解反应将与层状结构修复和表面重建同时发生。由于Mn4正极(0.67 A)和Co3正极(0.69A)的离子半径类似,Mn4正极可能会在内部扩散,优先占据Co位点。同时,硫脲的分解产物可以与LixCoO2反应,实现将SO42−聚阴离子和N元素掺杂到LCO晶格中,并在LCO表面原位涂层Li2SO4。最后,构建了具有目标化学计量、多元素掺杂和表面改性的理想构型,提高了再生LCO(即MNS-LCO)在高压下的性能。
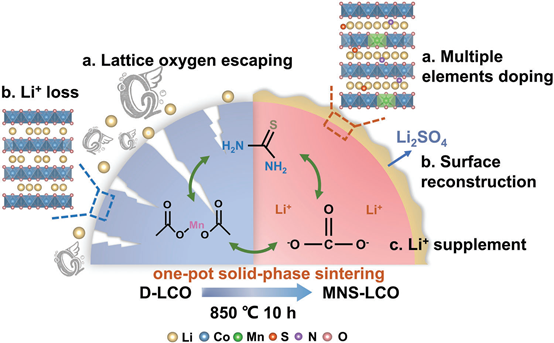
图1. D-LCO正极的上循环过程示意图
为了评价我们的正极再生过程的有效性,我们分析了上再生的LCO相对于D-LCO的变化。首先进行了场发射扫描电镜(FESEM)观察,研究了重建前后D-LCO的形貌变化。如图1a所示,经过长期循环后,D-LCO颗粒表面存在明显的微裂纹。此外,利用透射电镜(TEM)和高分辨率透射电子显微镜(HRTEM)进一步揭示了D-LCO和MNS-LCO之间的微观结构差异。如图1b、c所示,D-LCO的体结构保持了层状结构,平面间距为0.289和0.207 nm的明显晶格条纹分别对应于LCO的尖晶石结构(Co3O4)(220)平面和(104)平面,值得注意的是,D-LCO中出现了一些孔洞(图1b中的插图),Li正极的长期脱嵌入过程导致D-LCO晶体的结构坍塌。对于合成的MNS-LCO,结构缺陷得到了有效的修复,而相应的表面变得相对粗糙(图1d),表明表面重建成功。
此外,还可以清晰地观察到一个具有纳米尺度的超薄层(图1e)。从图1f中的HRTEM图像中,可以清楚地识别出与Li2SO4的(114)平面相关的0.170 nm的晶格条纹间隔,对应再生的MNS-LCO的表面重建。由图1g中的快速傅里叶变换(FFT)模式支持。值得注意的是,在图1h中上MNS-LCO中也观察到原始LCO(003)平面对应的晶格条纹间距为0.470 nm,这与FFT模式一致(图1i)。也就是说,Mn、N和S元素的共掺杂不影响LCO的体结构。图1j中的局部区域元素映射显示,Co、O、Mn、N、S元素均匀分布在上升后的MNS-LCO粒子中,验证了锰、N和S元素成功融入MNS-LCO的整体结构。结合以上分析,可以合理地推断,部分S参与了MNS-LCO粒子表面Li2SO4粒子的形成;而另一个S与N和Mn一起进入MNS-LCO的体结构。

图1.a)D-LCO的SEM图像;b,c)D-LCO在不同放大倍数下的HRTEM图像;d)MNS-LCO的SEM图像;e-i)MNS-LCO的HRTEM图像和相应的FFT模式;j)MNS-LCO的各种元素映射图像。
经过再生/修复后,由于Mn、N和S的共掺杂作用,MNS-LCO的(003)平面有轻微的向左偏移,而Co3O4的特征峰则消失了。因此,Mn、N和S元素的掺入导致了MNS-LCO沿c轴方向的间距的增加,促进了Li正极的扩散,增强了MNS-LCO正极的电化学性能。基于D-LCO、P-LCO和MNS-LCO样本的XRD模式,进一步进行Rietveld细化分析,以提供更多的定量结构信息(图2a,b)。因此,MNS-LCO的拟合c参数显著高于D-LCO和P-LCO,说明Mn、N和S元素的共掺杂扩大了层间距。根据重新定义的结构参数,Mn优先占据Co位点,而N、S掺杂剂倾向于占据Li-O板。因此,Li─O板中的N和S与最近的O紧密结合,产生了较强的S─O和N─O键。值得注意的是,由于MNS-LCO的Li2SO4含量较低,因此没有检测到与Li2SO4相关的衍射峰。
根据拟合参数,D-LCO含有36.23%的Co3O4;而在MNS-LCO中没有发现任何Co3O4衍射峰,这表明上升循环的MNS-LCO获得了良好的结构恢复。此外,利用拉曼光谱验证了D-LCO正极中Co3O4相的存在。如图2c所示,在D-LCO正极中,在700cm−1处的能带属于Co3O4。对于MNS-LCO样品,Co3O4的条带消失;在493和603cm−1处观察到两个特征峰,分别对应于LCO的O─Co─O弯曲模式(Eg)和Co─O拉伸模式(A1g)的晶格振动。得益于MNS-LCO中Mn、N和S元素的共掺杂效应,Eg和A1g模式的低波数有轻微的变化,表明与M-LCO和NSLCO相比,c轴晶格参数增加。从图2d的傅里叶变换红外光谱(FTIR)光谱中,1108cm−1波段显示了MNS-LCO样品中存在SO42−官能团,表明高价S掺杂到MNS-LCO中形成SO42−聚阴离子。此外,通过XPS深度进一步研究了Co、Mn、N、S在NMS-LCO中的分布(图2e-h)。随着蚀刻时间的延长,Co、Mn、N和S显示出几乎不变的信号强度,表明Mn、N和S被有效地掺杂到LCO的本体结构中,从表面到内部呈均匀分布。S元素不仅分布在MNS-LCO粒子表面形成Li2SO4,也掺杂到LCO的体积结构。
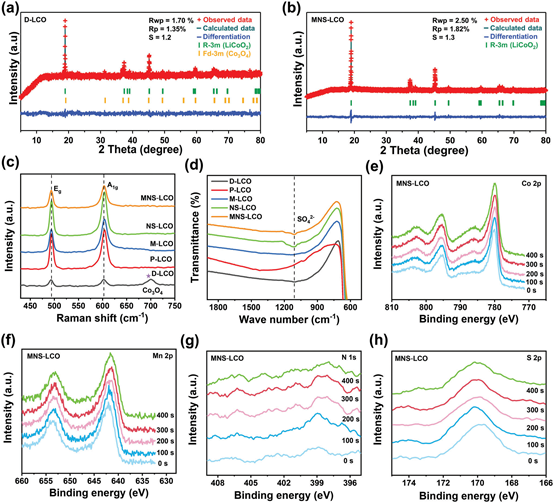
图2.a)D-LCO的XRD图精修;b)上循环MNS-LCO的XRD图精修;c)D-LCO、P-LCO、NS-LCO和MNS-LCO的拉曼光谱;d)D-LCO、P-LCO、M-LCO、NS-LCO和MNS-LCO的FTIR光谱;e) Co2p、f) Mn 2p、g) N 1s、h)MNS-LCO的S 2p峰随着正极蚀刻时间增加到400 s的XPS深度分布。
为了揭示在充电/放电过程中潜在的相变和动力学,利用循环伏安(CV)测量D-LCO和MNS-LCO在3.0−4.5V电压范围内的特性曲线,结果如图3a所示。显然,MNS-LCO作为Co3正极/Co4正极氧化还原偶,在4.04/3.86V处有一个明显的氧化还原峰。同时,由于LCO从六角形(H1)向单斜形(M)的可逆转变,在4.11/4.06 V和4.18/4.14 V处可以观察到两对弱峰。MNS-LCO相对于D-LCO的氧化还原峰更高,表明其电荷转移动力学更快。在4.5 V的充电电压下,D-LCO正极材料与MNS-LCO相比表现出明显的极化,说明MNS-LCO的可逆性优于D-LCO。
此外,MNS-LCO的氧化还原峰之间的迟滞率为0.18 V,小于D-LCO(0.26 V),说明MNS-LCO可能加速Li+在正极表面的扩散动力学。这种有利的现象可能是由于多元素掺杂的协同效应,它不仅抑制了氧化,而且削弱了充放电过程中的极化。再次,在dQ/dV曲线中进一步研究了循环过程中的相变行为。如图3b所示, MNS-LCO在3.93/3.89 V处有一对尖峰,在4.09/4.06和4.19/4.17 V处有两对弱峰,分别对应于Li0.5CoO2附近成分的一阶金属-绝缘体跃迁和有序-无序跃迁。然而,在D-LCO中,只观察到在3.92/3.88 V处的一对峰,表明其可逆性较差。以上结果证实,在MNS-LCO表面添加Li2SO4涂层Li,并与微量的Mn、N、S共掺杂,可以稳定LCO的层状结构,提高高压下氧的稳定性。此外,在第一个循环中,在0.2 C的3.0~4.5V的电压范围内,比较了各种正极材料的充放电曲线。如图3c所示,MNS-LCO的初始放电容量为188.7mAhg−1,高于D-LCO、P-LCO、M-LCO和NS-LCO(分别为109.4、174.4、169.7和168.8 mAhg−1)。同时,MNS-LCO(3.9 V)的放电平台也高于其他正极材料。从倍率能力(图3d)来看,在不同的电流密度,MNS-LCO的倍率性能优于D-LCO和P-LCO,这是由于加快电荷转移动力学,在大电流密度(5 C)下,容量保持了144.8mAhg−1。
可见,MNSLCO的最高放电容量在0.1 C、0.2C、0.5 C、1C、2C、3C和5C下分别为为188.1,188.2,183.7、176.3、165.3, 157.3, and 144.8 mAh g−1。D-LCO、P-LCO、M-LCO、NS-LCO和MNS-LCO在0.5 C、3.0−4.5V电压范围下的循环性能也见图3e。值得注意的是,M-LCO、NS-LCO和MNS-LCO的稳定性明显优于D-LCO。同时,D-LCO、P-LCO、MLCO、NS-LCO和MNS-LCO的保留率分别为66.1%、86.6%、96.4%、98.5%和92.5%。虽然NS-LCO具有最高的保留率,但由于多元素掺杂的协同效应,MNS-LCO在100次循环后保持了最高的放电能力。
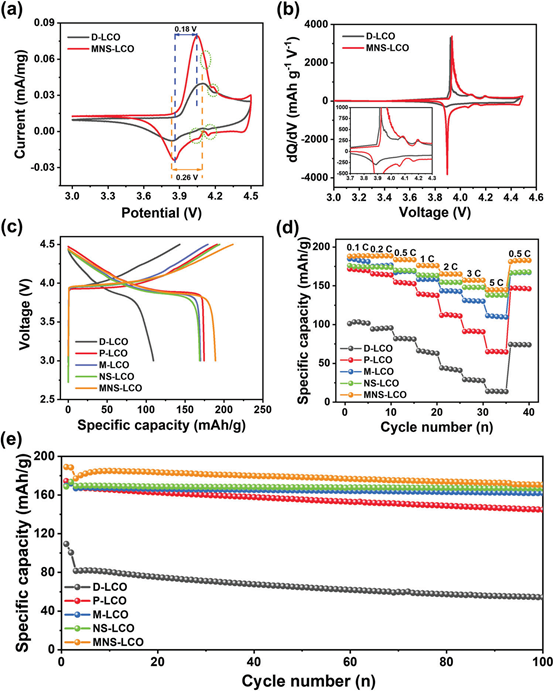
图3.a)D-LCO和MNS-LCO在0.1 mV S−1扫描速率下的CV曲线;b)0.1C下D-LCO和MNS-LCO在0.1 C时的dQ/dV曲线;c)0.2C下D-LCO、P-LCO、M-LCO和MNS-LCO的第一充放电曲线;d)3.0和4.5 V不同电流密度下的倍率性能;e) D-LCO、P-LCO、M-LCO、NS-LCO和MNS-LCO在0.5 C下循环性能。
为了探究MNS-LCO在高截止电压下的潜在高稳定性机制,采用原位XRD测量来监测从0.2 C的第一次开路电压(OCV)到4.5 V的相变。考虑到相变的优良灵敏度,我们选择了(003)峰来揭示电化学过程中晶格常数c的变化(图4a-d)。随着Li+离子在开始时的逐渐脱插(电压高达4.2 V),由于氧层之间的斥力,MNS-LCO的(003)峰迁移到低角度,其中MNS-LCO的晶体结构可能发生从H1向M的转变,这与CV和dQ/dV的结果一致。当充电到高压(例如,4.45 V)时,(003)峰迅速向右移动,这是由于缺乏剩余的锂支撑而导致锂层的收缩。说明MNS-LCO经历了从M到O3的相变。与D-LCO相比,MNS-LCO在(003)峰的强度和位置上表现出更平滑的演化,这表明在MNS-LCO中都有效地缓解了绝缘体-金属相变和M相变。同时,MNS-LCO中(003)峰的变化相对较弱(ca。0.48°);而D-LCO相应的变化幅度为0.51°,说明MNS-LCO内的剧烈体积变化被抑制,不可逆相变减弱。与D-LCO中(003)峰位置的显著变化不同,MNS-LCO的(003)峰在完成初始充放电循环后可以移动到原始位置。
MNS-LCO具有良好的结构可逆性,可以归因于N、S和O之间存在强共价键,以及Li2SO4在LCO表面的涂层,有利于提高层状结构的稳定性。同时,在LCO粒子的锂层中掺杂N和S,并有效地引入Co层,可以在不破坏晶体结构的情况下承受大体积变化,从而提高了电化学性能。进一步,用电化学阻抗谱(EIS)研究了充放电过程中界面反应动力学的演化。如图4e所示,D-LCO和MNS-LCO的初始状态下的电荷转移电阻(Rct)值分别拟合为323和298 Ω。其中,Li+在MNS-LCO中的最佳扩散可归因于Mn、N和S共掺杂引起的层间距扩大,以及快速离子导体Li2SO4在表面的涂层。当充电到4.5V时,D-LCO的Rct迅速上升到547.9 Ω,远远高于MNS-LCO(99.5Ω)。放电至3.0 V后,MNS-LCO和D-LCO的Rct值分别为29.8和137.3 Ω,说明MNS-LCO即使在高损耗状态下仍能保持电极/电解质界面的稳定性;而D-LCO则发生退化反应。
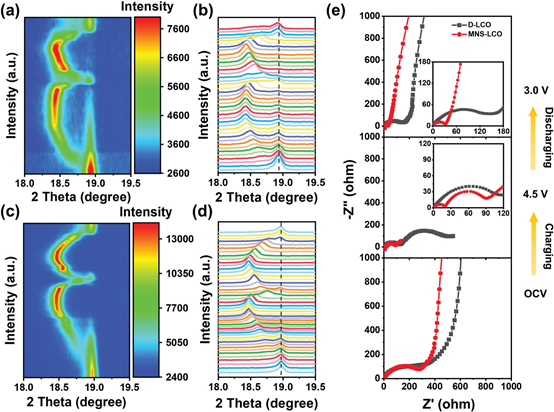
图4. 在0.2 C 电压范围为4.5-3.0V的初始充/放电过程中,D-LCO a-b)和MNS-LCOa-b)的原位XRD结构演化;e) D-LCO和MNS-LCO在初始循环后OCV、4.5和3.0 V处的EIS阻抗谱图
为了监测D-LCO和MNS-LCO中Co价态和晶格O的转变,采用XPS检测不同充电阶段后的化学键变化。从图5a可以明显看出,D-LCO的Co2p3/2特征峰在充电时从779.8显著转移到780.8,说明在充电过程中Co4+在过渡金属层中不断积累。相比之下,Co2p特征峰值MNS-LCO显示一个小转变,进一步证明表明,N,S和锰的共掺杂不仅可以作为一个“支柱”的层结构,还调节高价的电子配置Co4+/O−,高压有助于抑制晶格氧的价振荡和退化。如图5b所示,O 1s分为529.7(蓝色区域)、531.6(粉色区域)和532.8eV(橙色区域)三个峰,分别为晶格氧(晶体网络中的氧原子特征)、氧空位(表面吸附氧或羟基)和化学吸附氧(弱吸附分子水)。值得注意的是,N─O(532.3eV)和S─O(532.2eV)键仅在MNS-LCO样品中发现(图5c),说明N,S在MNS-LCO中有效掺杂。
此外,D-LCO和MNS-LCO样品的晶格O在充电至4.5V后的峰值强度均下降,表明电极材料和电解质之间形成了正极电解质界面(CEI)层。与MNS-LCO相比,D-LCO在充电到4.5V或循环100个循环时,晶格氧的峰值强度更明显地降低。此外,在100个循环后,D-LCO中O 1s的峰值位置发生了负偏移(图5d),表明其结合能明显减弱。以上结果充分证明,Mn、N、S元素的共掺杂可以消除晶格O的剧烈振荡,抑制晶格O对高压下电荷补偿的参与。
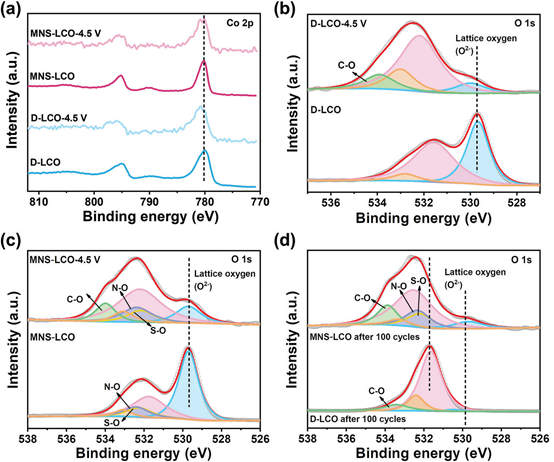
图5. 非原位XPS:a) Co 2p光谱,b)D-LCO在原始和4.5V下的O1s光谱,c)MNS-LCO在原始和4.5V的O1s光谱,d)D-LCO和MNS-LCO在100个循环后O1s光谱。
为了阐明Mn、N和S共掺杂后电化学性能提高的机理,我们在不同的充电状态下进行了DFT计算。将六方LCO单元的平衡晶格常数优化为a=b=2.848Å,c=14.042Å。随后的计算都是基于一个135个原子的超级单体。Li0.2CoO2通过随机去除43个Li原子来模拟,如图6a所示。Mn、N、S掺杂LCO的优化结构如图6b所示。结果表明,原始LCO的投影态密度(PDOS)是非磁性的,图6c中Co3+处于低自旋态(S = 0)。在释放后,晶体场被打破,从而产生磁态。进一步计算了锰掺杂在Co位和Li位的生成能。
结果表明,Mn掺杂在Co上的生成能为0.22 eV,低于在Li位上的生成能(0.32eV),说明Mn更倾向于占据Co位。更重要的是,Mn的掺杂也使主要的O-p态远远低于费米能级。在N和S占据Li层间隙位置的情况下,形成能最低,为1.62 eV,突出了结合XRD的Rietveld细化分析,将N、S掺杂到Li-O板中的可行性。与LCO的PDOS相比,N、S的掺杂可以减小MNSLCO的带隙。从图6d可以看出,MNS-LCO的PDOS可以清楚地看出,Mn、N和S的共掺杂可以减少带隙,进一步使主要的O-p态远低于费米能级,有利于抑制LCO的氧演化。值得注意的是,Co-d、Mn-d、N-p和O-p的自旋向下态穿过费米能级,促进了MNS-LCO在充放电过程中的电子转移。由此可见,Mn在MNS-LCO材料中占据了Co位点,而N和S被掺杂到Li层的间隙位点中。
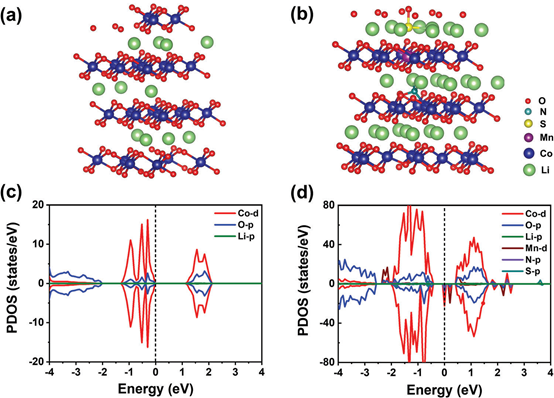
图6. a) Li0.2CoO2、b) Mn、N、S掺杂LiCoO2的优化结构。PDOS c) LCO和d) MNS-LCO。
总结与展望
在这篇文章中,作者成功地演示了一种无损的一锅工艺,使D-LCO直接转化为高能量密度的MNS-LCO正极材料,具有易于放大的潜力。基于所开发的三合一固相烧结策略,可以有效恢复D-LCO的组成和晶体结构,同时实现Li2SO4原位表面涂层和Mn掺杂Co位,N、S掺杂Li-O板。所有这些协同效应都是被证明能有效减少O2逃逸,抑制有害相变,稳定重建LCO的层状结构。因此, MNS-LCO正极材料表现出优越的电化学性能,包括高比放电容量、优异的速率能力和在4.5 V的高截止电压下令人满意的循环稳定性,优于D-LCO和新商用的LCO。该策略不仅为D-LCO的回收和上循环为高压LCO提供了一个很有前途的解决方案,而且还可以扩展到将其他降解的正极材料升级为高性能锂离子电池,这将加速回收行业的可持续发展。
审核编辑:刘清
-
三合一PMU CN8914用于智能电表,实现小尺寸解决方案2025-02-21 2850
-
聚阴离子锚定策略提升高电压LiCoO2的反应动力学性能于EES中探究2024-11-14 2096
-
三合一筋膜枪功能说明2024-01-12 1278
-
高性价比三合一移动电源设计技术2023-11-08 582
-
三合一智能音箱演示2023-03-15 662
-
移动电源三合一方案都有哪些_移动电源三合一方案哪种最稳定2017-12-29 18667
-
新人提问:移动电源方案软件三合一好还是硬件三合一好?2015-10-23 4328
-
移动电源硬件三合一方案和软件三合一方案对比2013-10-21 5585
-
同步整流 移动电源三合一问题2013-09-18 9780
-
移动电源方案究竟硬件三合一还是软件三合一?2013-09-15 14663
-
LabVIEW边干边学系列三合一2012-08-12 3122
全部0条评论

快来发表一下你的评论吧 !
