

N杂五元芳烃的非共价成键机制研究
描述
01 引言
N杂五元芳烃在生物和药物化学中占有重要地位,它是药物分子的基础结构单元,也是蛋白质、辅酶、生物碱、DNA 等生物分子的核心骨架。由该基团参与的非共价相互作用(NCI)会介导分子识别、结合、催化和药物传递等过程,从而在调节生物分子结构和诸多生命过程(如光合作用、信息存储、DNA 复制方面)中发挥着关键作用。因此,对N杂五元芳烃的非共价相互作用及其本质特征进行探究,有助于阐明许多复杂生物事件的物理化学机制。因此,本项目选取噻唑作为模型N杂五元芳香杂环化合物,采用高分辨转动光谱结合理论计算方法精确解析其与四氟化碳、六氟化硫的非共价相互作用的拓扑几何结构、结合强度和相互作用的本质。
02 成果简介
本项工作,基于密度泛函理论对噻唑-CF4 和噻唑-SF6体系进行了几何结构优化和振动频率分析,进而在转动光谱实验中成功地指定和分配了属于各自最稳定构象的转动谱线,并结合量子化学理论计算阐明其分子间的非共价成键机制。实验和理论计算的结果证实;所观察到的噻唑-CF4异构体主要是由位于噻唑环平面的CF4的碳原子通过N···CCF4相互作用形成的,且每条转动跃迁表现出由-CF3顶部内旋引起的A/E扭转分裂;而噻唑和SF6主要通过范德华相互作用来稳定,SF6位于噻唑环上方。虽然静电和色散都是噻唑-CF4和噻唑-SF6二聚体形成的主要因素,但在噻唑-SF6复合物中,色散项显得尤其重要。另外,在比较几种不同理论水平的计算精准度后发现,相较于M062X和PBE方法,B3LYP方法对噻唑-CF4复合物的计算更为准确,但对于噻唑-SF6复合物来说,ωB97XD方法得到的理论值的误差更小。
03 图表导读
(a)
(b)
(c)
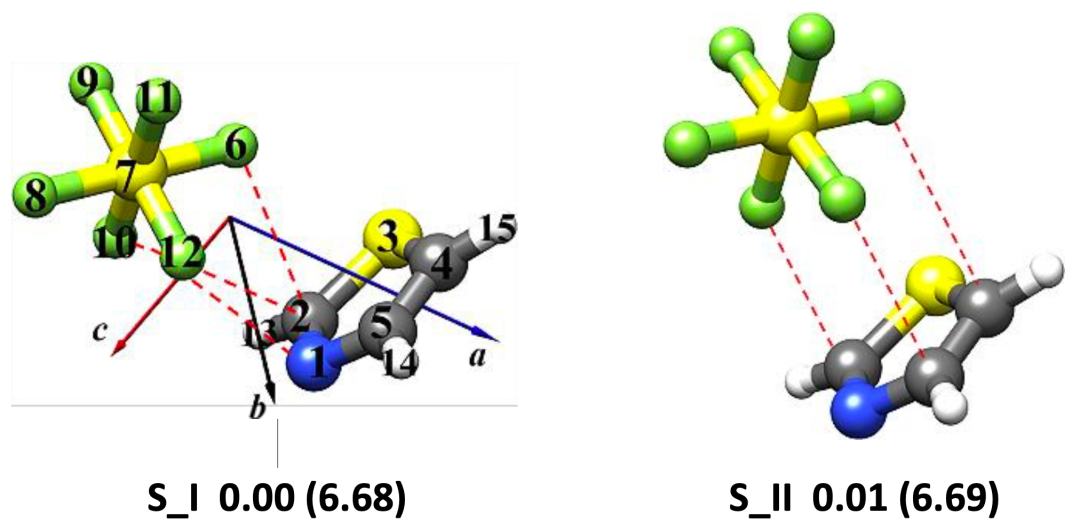
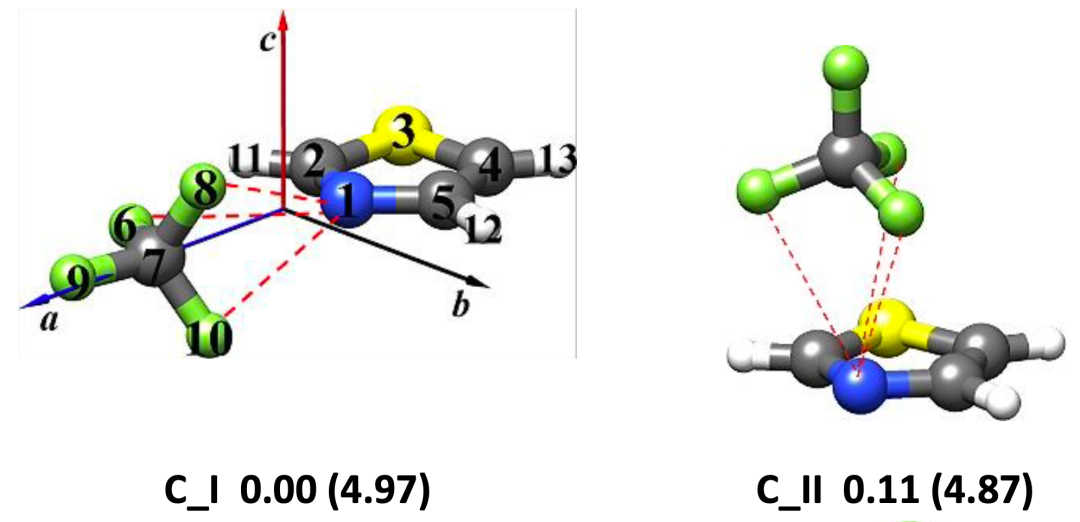
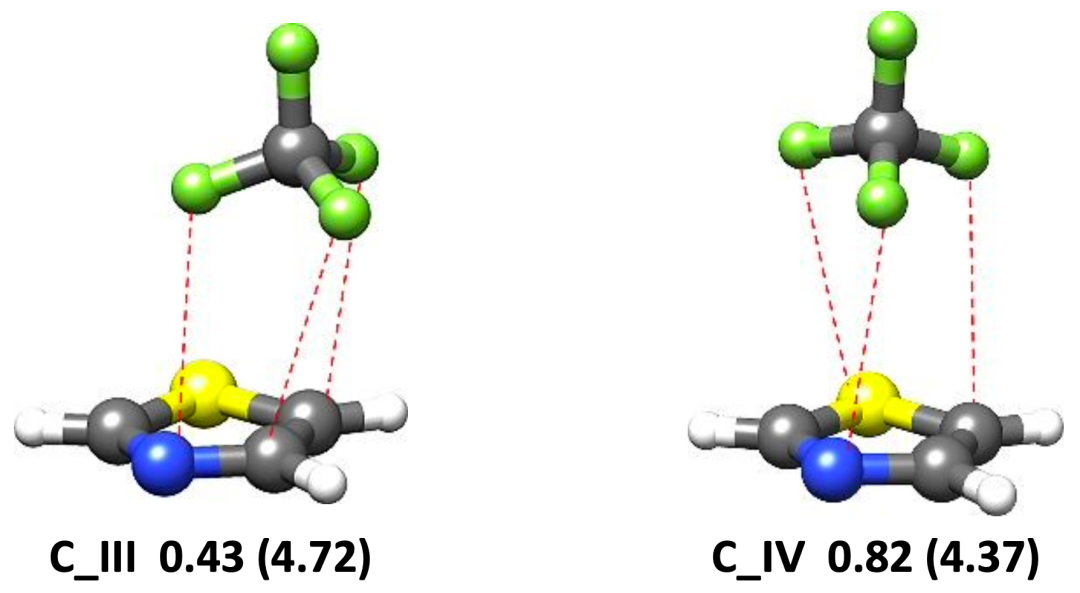
图 1. 理论计算得到的噻唑-CF4 (a)和噻唑-SF6 (b)团簇的稳定构象
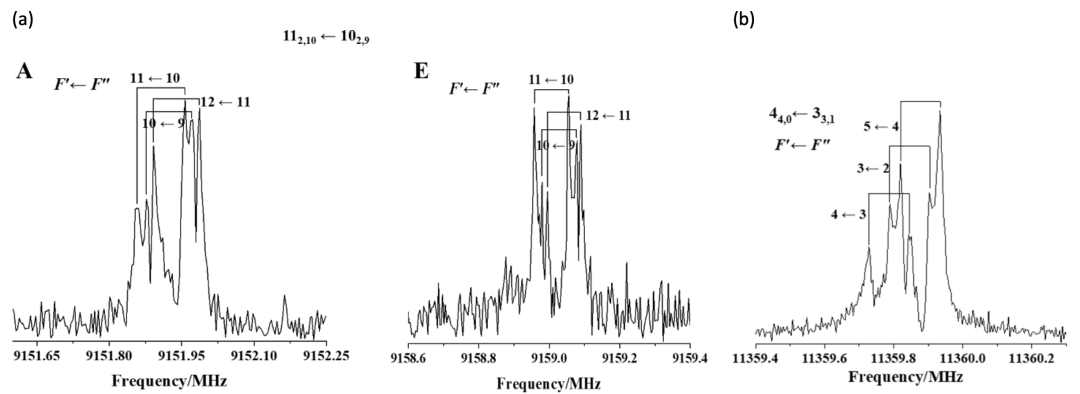
图2.(a) thiazole-CF4的部分转动光谱图;(b) thiazole-SF6的部分转动光谱图
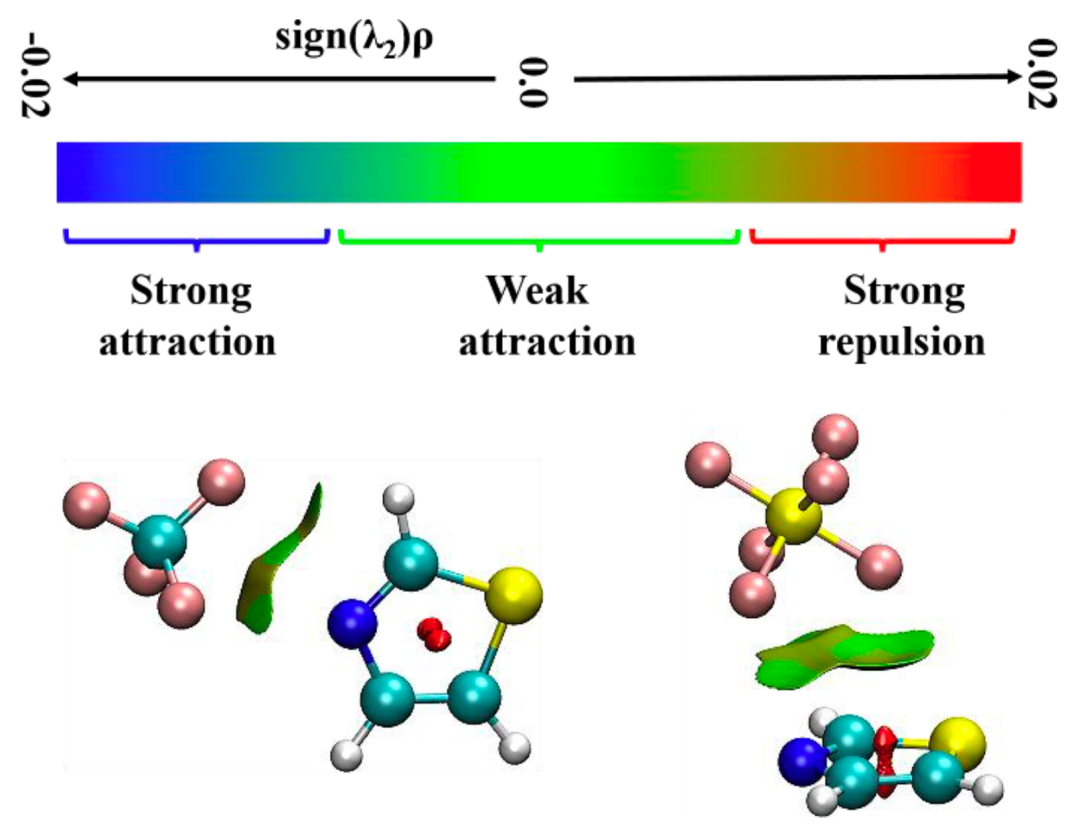
图3.噻唑-CF4和噻唑-SF6团簇的簇内分子间非共价相互作用
表1.不同理论方法计算得到的thiazole-CF4的光谱参数
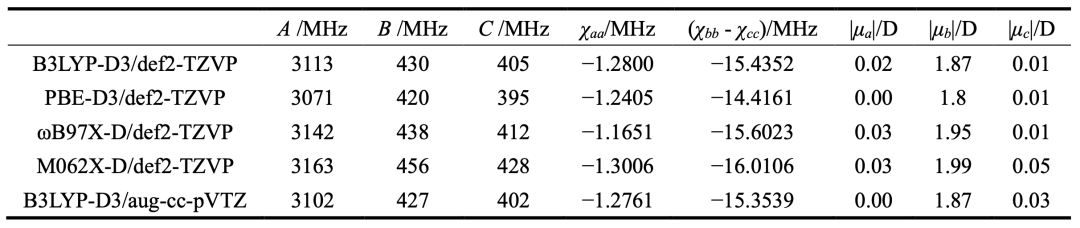
表2.不同理论方法计算得到的thiazole-SF6的光谱参数
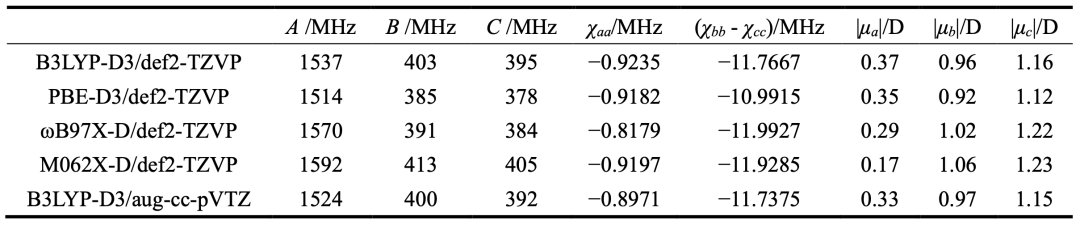
表3.实验测定thiazole-CF4和thiazole-SF6的光谱参数

表4.thiazole-CF4和thiazole-SF6的SAPT分析的结果(kJ mol−1)
04 小结
本文使用鸿之微 Device Studio平台搭载的BDF程序开展有关噻唑-CF4和噻唑-SF6体系的量子化学理论计算,再利用傅里叶变换微波光谱技术获取各自最稳定构象的转动光谱数据,从而确定分子结构并揭示非共价成键机制。研究表明,噻唑-CF4最稳定的构型是由静电和色散主导的CF3···N相互作用拓扑,其 rN···C距离为3.407 (2)Å;而位于噻唑环上方SF6是由范德华相互作用与噻唑结合,色散是主要吸引项。在理论计算方面发现,B3LYP-D3(BJ)/def2-TZVP 理论水平能够为噻唑-CF4 和噻唑-SF6复合物提供与实验相近的转动常数值,且ωB97X-D3/def2-TZVP方法略微提高了噻唑-SF6的预测精度。
审核编辑:刘清
-
Java非阻塞通信研究2009-08-10 367
-
嵌入式网络终端报文收发机制研究与实现2009-09-11 742
-
DDS频谱杂散分析及其抑制研究2010-10-20 510
-
共价晶体结构2009-08-06 8472
-
新大管道杂散电流干扰影响研究2015-11-16 664
-
基于非联合型学习机制的学习神经元模型2017-11-29 1952
-
苹果正研究iPad键盘盒的铰链机制,提供可调节触觉键反馈2020-09-25 2326
-
介绍一种光触发的新型OER机制COM2023-03-20 2732
-
弱相互作用对有机光电性质调控的理论研究2023-07-31 1831
全部0条评论

快来发表一下你的评论吧 !
