

锂离子电池表面改性正极的快速嵌锂机理
描述
研究背景
利用氧化材料(如Li2ZrO3)对正极表面进行改性,锂电池正极的循环稳定性和倍率性能得到显著提高。部分研究指出,改性层的成分通过与电解质发生电化学反应而改变,促进离子导电CEI层的形成,为进一步了解电池运行下界面结构的变化,以及改性正极-有机电解质界面上锂(脱)嵌入的反应速率,必须使用单一的技术来研究电池反应过程中从电极侧到电解质侧的界面结构变化。
成果简介
日本东京工业大学Masaaki Hirayama教授利用原位NR研究Li2ZrO3改性LiCoO2正极和未改性正极,发现表面改性有利于形成致密的、由无机物种组成的CEI,未改性的表面覆盖着相对稀疏且浸渍了电解液的CEI。研究发现,锂在插层过程中的脱溶主要发生在CEI和LiCoO2表面,锂在CEI上的快速溶解可能有助于表面改性的正极具有优异的倍率性能,提出了一种通过利用低离子导电性氧化物进行表面改性来实现快速插层的机制。该工作以“Fast Lithium Intercalation Mechanism on Surface-Modified Cathodes for Lithium-Ion Batteries”为题发表在Advance Energy Materials上。
研究亮点
(1)用PLD方法在SrRuO3(100)/SrTiO3(100)衬底上外延生长了LiCoO2(104)和Li2ZrO3改性的LiCoO2(104)薄膜。
(2)与未改性的LiCoO2(104)相比,改性的LiCoO2具有更好的锂插层速率能力。
(3)改性表面的内层由无机物质组成,未改性的表面被相对稀疏的电解质浸渍的CEI覆盖,与LiCoO2表面相比,改性LiCoO2优越的速率能力可能源于CEI上锂能更快脱溶,并稳定电极表面,为适合大功率工作的正极-液-电解质界面设计提供了一种新的设计原则。
(4)利用原位NR直接观察循环过程中的界面结构,能够同时检测化学成分和形态信息,是阐明电池反应详细机理的有力方法。
图文导读
图1为在SrRuO3(100)/Nb:SrTiO3(100)上合成的未改性和Li2ZrO3改性的LiCoO2(104)薄膜的XRD图和XRR谱图。层状岩盐型LiCoO2沿SrTiO3<100>、<010>和<011>方向分别有104、01-4和01-8衍射峰。这证实了未改性和Li2ZrO3改性的LiCoO2薄膜的外延生长。Li2ZrO3沉积过程中没有形成固溶体,这归因于Zr4+在层状岩盐型结构中的溶解度差。未改性和改性的LiCoO2膜的厚度分别为22.1 nm和22.0 nm, SLD值分别为38.1×10−4和38.3×10−4 nm,与XRD结果一致,表明未形成LiCoO2-Li2ZrO3固溶体,两种样品的表面粗糙度均为≈1 nm,表面面积无显著差异。因此,未改性和Li2ZrO3改性的LiCoO2(104)薄膜适合研究改性对正极-电解质界面电化学反应的影响。
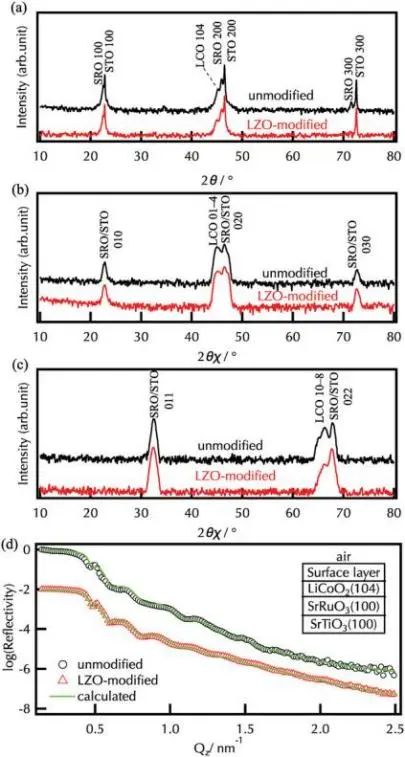
图1. a) SrRuO3(100)/Nb:SrTiO3(100)上未改性和Li2ZrO3改性的LiCoO2膜的a)面外、b,c) 面内XRD谱图和d) XRR谱图。LCO、LZO、SRO、STO分别代表LiCoO2、Li2ZrO3、SrRuO3、SrTiO3。拟合模型和计算得到的XRR谱如图1d所示。
在100C(300μA cm−2)条件下,两种膜的放电容量相近,约为125 mAh g−1,相当于LiCoO2的可逆锂插层。高容量保持是因为电子和锂离子在26 nm厚的薄膜电极内的传输距离较短。未改性薄膜的Vave在第一次循环时放电电压为3.53 V,到第五次放电时Vave值逐渐下降到3.49 V,这是由于放电结束时电压降低,在之后循环中 Vave 值随着电流密度降低而增加。相反,改性薄膜的放电电压较高,达到3.80 V,在100C下的五个循环中没有明显下降。XRD分析证实,未改性和改性薄膜的晶体结构在生长状态下没有发生实质性变化,此外,在20次充放电循环过程中,高度可逆的插层容量表明26 nm厚的钴酸锂表面没有发生实质性退化。因此,与未改性的薄膜/电解质界面相比,Li2ZrO3改性的LiCoO2/电解液界面表现出更高的锂插层反应速率。
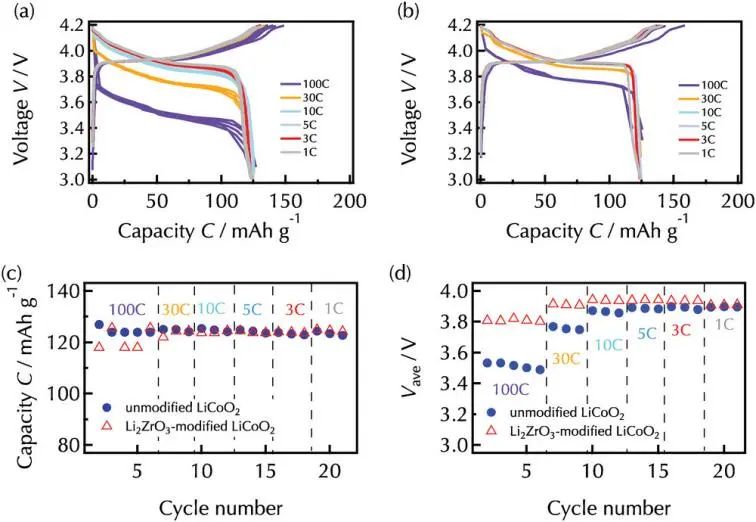
图2. a)未改性LiCoO2和b)Li2ZrO3改性LiCoO2在100C(300μA cm−2)到1 C(3μA cm−2)不同的放电电流密度下充放电曲线,充电电流密度固定为1C。c)放电容量的变化和 d)从(a、b)中获得的平均放电电压。在100C下改性LiCoO2放电容量的变化是由于充放电设备的时间分辨率所致。
图3是两种薄膜在第二个周期中不同放电电压下的EIS图。对于这两种薄膜,在4.1V、4.0V和3.9V都有明显的扭曲半圆,其直径从4.0增加到3.9V。在3.7V和3.5V时,10 Hz以下的低频区阻抗值急剧增加,而在10Hz以上观察到的半圆比例随着电压的变化而相对保持不变。这表明,3.9V以上的扭曲半圆由两个时间常数相近的分量组成。因此,作者使用具有RLFCPELF和RHFCPEHF两个RC分量的等效电路来改进Nyquist曲线图。
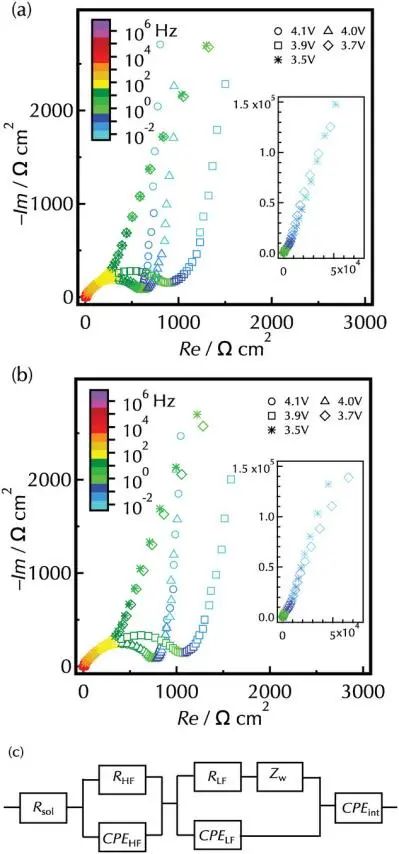
图3. a)未改性的LiCoO2和 b)Li2ZrO3改性的LiCoO2薄膜在第二个循环中放电至不同电压后的奈奎斯特图。c) 采用等效电路进行频谱拟合。
在第二次充放电循环期间,未改性膜和改性膜的RLF值在充电时从3.9V下降到4.1V,放电时从4.1V可逆地增加到3.9V。由于(脱)嵌锂速率会随着正极材料中Li+浓度的变化而变化,因此RLF与正极-电解质界面上锂(脱)插锂的界面电阻有关。与RLF相比,两种薄膜的RHF值随电池电压的变化而保持相对稳定,考虑到电解液和锂正极中包括的电化学过程中的微小电阻,正极侧可以包括另一个具有高时间常数的电阻分量,如果假定Li2ZrO3基CEI层的离子电导率与Li2ZrO3相当,则计算出CEI层的锂转移电阻为≈15Ω cm2,这导致在100C工作时极化为5 mV,RHF不能确定为RCEI。因此,在高频下,电子接触电阻Re可能是半圆的主要电阻成分。总体而言,两种薄膜在所有条件下的 RLF 值都高于RHF值,这意味着在薄膜电池中,正极界面的锂插层是决定速率的步骤。正极-CEI-电解质界面包含了锂嵌入的几个基本过程:i)溶剂化锂在CEI表面的吸附和脱溶,ii)锂在CEI层中的转移,以及iii)锂嵌入伴随着正极-CEI界面的电子转移。过程(i)和(ii)的速率可能取决于CEI层的化学成分和结构,这些成分和结构会因表面改性而改变。锂嵌入过程的速率(iii)取决于反应速率常数和正极和电解质两侧界面处的锂浓度,作者假设未改性和改性的LiCoO2薄膜表面没有明显的劣化,因为在4.2V和3.0V电压区域循环过程中充放电容量保持一致。这表明锂嵌入过程的速率主要取决于电解质侧界面。
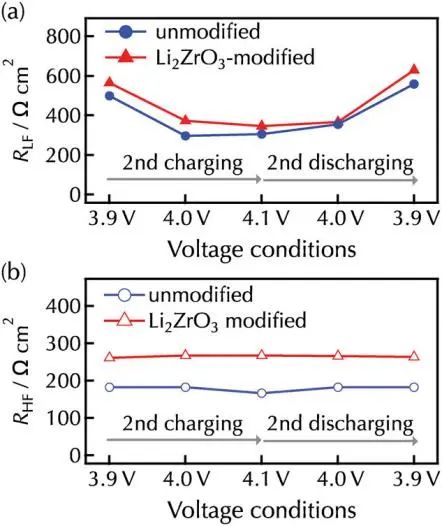
图4. 未改性和li2ZrO3改性的LiCoO2在第二次充放电时的a) RLF和b) RHF值的变化
图5为未改性和Li2ZrO3改性的LiCoO2(104)薄膜在第三次放电后的NR谱拟合结果和nSLD曲线,与原始薄膜相比,由layer1/layer2/LiCoO2/SrRuO3/SrTiO3组成的双表面层模型与两种薄膜的反射率曲线非常吻合。未改性和改性的LiCoO2层均表现出相似的nSLD和粗糙度,这表明未改性和改性的LiCoO2之间没有明显的结构差异。对于未改性的LiCoO2,外层和内层的厚度分别为2.2nm 和1.5nm,改性后的LiCoO2包覆了厚度为1.5nm的外层和内层,nSLD分别为3.09×10−4nm−2和2.42×10−4 nm−2,据报道,原始状态下的表面层被主要由电解质物质分解形成的CEI物质所取代,考虑到CEI成分的nSLD 值,外表层主要由Li2ZrO3和 CH2OCO2Li组成,底表层主要由 LiF、Li3POxFy和 LiOH组成。改性表面内层的nSLD值略高于未改性表面,可能是由于基于Li2ZrO3的成分,如 Li2ZrOxFy。双表面层模型与先前的研究一致,表明无机CEI物质最初在正极表面形成,然后在正极表面堆积有机CEI物质。NR和XPS分析均检测到未改性和改性LiCoO2表面之间CEI层的成分或厚度没有显著变化。
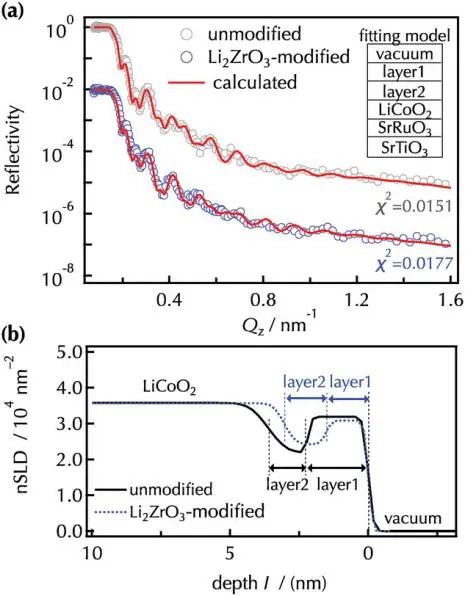
图5. a)观察并计算第三次放电后未改性LiCoO2和Li2ZrO3改性LiCoO2的NR光谱;b)细化了未改性和Li2ZrO3改性LiCoO2薄膜的nSLD曲线。采用SrRuO3/LiCoO2/layer2/layer1双表面层模型计算非原位NR光谱.
为消除采样过程对实验结果造成否影响,作者进行了原位NR分析,以探索初始反应过程中的电极-电解质界面。图6和图7显示了电池构建后以及在第三个循环中充电和放电状态下未改性和Li2ZrO3改性的LiCoO2(104)薄膜的原位NR拟合结果和nSLD曲线。与非原位NR分析一致,双界面层模型(SrRuO3/LiCoO2/内层/外层/电解液)与两个样品的反射率光谱密切相关。4.2V的带电状态下,未改性和改性的LiCoO2层的nSLD值高于电池构建后的观察结果,这对应于缺锂相Li1−xCoO2的形成,这可能是由于充电过程中锂的脱嵌作用。在放电状态下,nSLD值可逆地减小,证实了锂嵌入到Li1−xCoO2中,在两种薄膜的充放电循环过程中,LiCoO2层(≈分别为25 nm和1 nm)的厚度和粗糙度保持稳定,只有很小的变化。这些发现表明,可逆的锂(脱)插层发生在不降解LiCoO2相的情况下,与在充放电曲线中观察到的高容量保留一致。
电池构建后,在未改性的LiCoO2表面形成了厚度为2.6nm的内层,在充放电过程中,没有观察到明显的厚度、nSLD 或粗糙度变化,这表明,浸入电解液后形成的内层在随后的过程中保持电化学稳定。以前的研究表明,LiOH和Li2ZrO3等表面物种与电解液中的HF和H3PO4等杂质发生化学作用,生成LiF和Li3PO4,从而形成无机CEI层。然而,第三次放电后观察到的nSLD值远远高于理论值,电解液的nSLD值为5.67×10−4 nm−2时,可能会使nSLD值增大,因为电解液可能会填满CEI层内的空洞。因此,在未经改性的表面上形成的内层可能既包括无机CEI物种,也包括液体电解液。在Li2ZrO3改性的LiCoO2表面,电池的内层厚度在第三次放电后变化很小,这表明内部的CEI是在浸泡在电解液中后形成的,这是由于Li2ZrO3与电解液物质之间发生了化学反应。nSLD值显著低于未改性LiCoO2,且与非原位NR测得的改性LiCoO2的内部CEI值吻合,表明改性的Li2ZrO3层促进了致密的无机CEI的形成,孔洞相对较少,在电化学条件下,这种致密的CEI有助于液体电解液向内层的渗透率。因此,原位NR分析表明,表面改性既影响CEI层的密度,也影响液体电解液的浸润量,这些可能会改变锂离子在插层过程中的界面扩散过程。
在未改性和Li2ZrO3改性的LiCoO2中观察到明显的外层,厚度超过5nm,在电化学条件下,外层表现出不同于在干燥条件下观察到的不同成分和形态。未改性的LiCoO2表面外层在充放电过程化学成分发生了变化,相反,改性LiCoO2的外层显示出更低的循环前nSLD值,为3.46×10−4 nm−2,在4.2V时,nSLD值增加到3.69×10−4 nm−2,而在3.2V时,nSLD值基本不变,与内层类似,改性前后外层界面结构的变化不同。作者提出了三种可能的界面模型来解释外层倾斜的nSLD分布:i)具有成分梯度的CEI层,ii)电解液中离子浓度的分布,以及iii)固体CEI与液体电解质比率的深度变化。在模型(III)的基础上,作者探讨了充电和放电过程中外层的结构变化。对于未改性的LiCoO2,当充电到4.2V时,nSLD增加,表明在外层形成了有机CEI,放电3.2V时nSLD的减少表明有机物种的分解或去除。Li2ZrO3改性的LiCoO2表现出比未改性的变体更低的nSLD,这表明在外层有更高的无机/有机CEI比和液体电解液,值得注意的是,外层在充放电过程中nSLD和厚度没有显著变化,模拟了内层的行为。在充放电的初始阶段,改性LiCoO2的界面结构建立了一定的化学成分和形态。致密的内部CEI阻碍了LiCoO2与液体电解液之间的电子接触,从而阻碍了电解液物种的进一步分解,从而扩大了CEI层。相反,未改性的LiCoO2形成的内部CEI密度较低,导致循环过程中外部CEI的生长和分解。
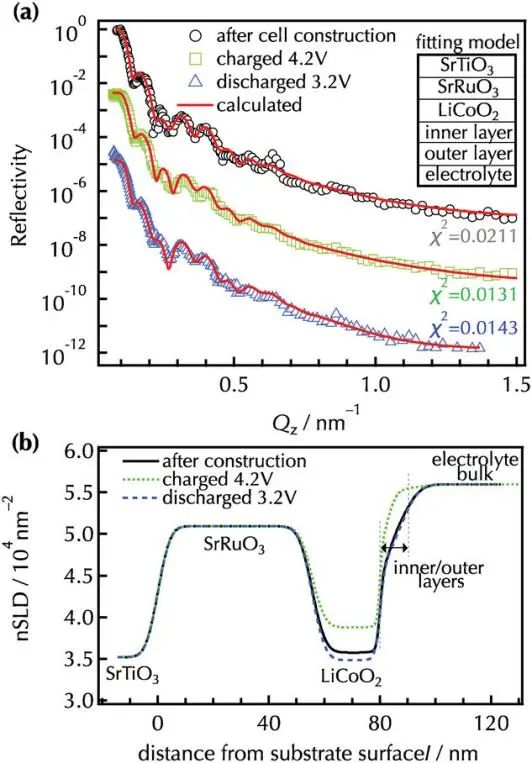
图6. a)观察和模拟了未改性LiCoO2薄膜的NR谱,b)改进了电池构建(3.3V)和充电到4.2V后第三次循环放电到3.2V后的nSLD曲线。用SrRuO3/LiCoO2/内/外层四层模型计算了原位NR光谱.
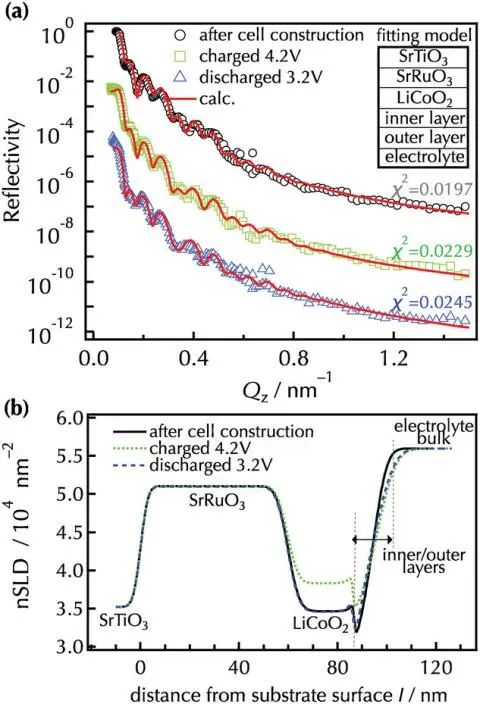
图7. a)观测和模拟了Li2ZrO3改性的LiCoO2薄膜的NR谱和b)精化的nSLD谱。用SrRuO3/LiCoO2/内/外层四层模型计算原位NR光谱.
对于这两种类型的LiCoO2,根据从LiCoO2表面到电解液本体的距离,界面结构分为内部和外部。内层和外层分别由无机CEI和有机CEI和电解液的混合物组成。Li2ZrO3的表面改性有利于无机CEI的形成,导致内层有致密的无机CEI,外层有较高的无机/有机CEI比值。基于这些关于界面结构的发现,作者预测锂插层过程中的锂转移过程如下:
未改性的LiCoO2:内层和外层的空腔浸渍有液态电解质。锂离子可在电解液中扩散,电解液的锂传导性大大高于CEI元件。锂离子从电解液快速穿过界面层,到达钴酸锂。然后离子脱溶,被钴酸锂表面吸收,接着插层进入钴酸锂晶格中。
Li2ZrO3改性LiCoO2:锂离子通过液体电解液扩散,浸渍在外层,到达内表面。由于内部CEI的高密度,大多数锂离子必须去溶才能扩散到内层。脱溶的锂离子通过LiCoO2和CEI内部的固-固界面嵌入到LiCoO2晶格中。
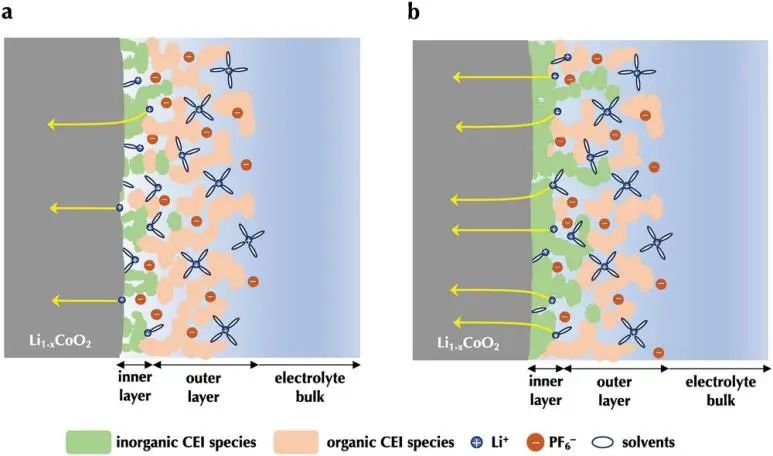
图8. a)未改性和b)Li2ZrO3改性的LiCoO2在放电条件下的电极/电解液界面示意图。
总结与展望
在改性表面的内层是由无机物种组成的,这有利于锂在内层CEI上的插层脱溶。相反,未改性的表面覆盖着相对稀疏且浸渍了电解液的CEI,脱溶主要在LiCoO2表面进行。因此,与LiCoO2表面相比,改性LiCoO2优异倍率能力可能源于CEI上锂的脱溶速度更快,而这种变化是无法在原位跟踪的。然而,这些变化应该与循环开始时的界面结构有关。作者强调了表面改性在促进锂离子快速解溶的界面结构方面所起的以前未明确的作用,从而导致更高的锂插层速率。这一发现将为表面改性材料的设计提供新的概念。
-
锂离子电池黏结剂2013-05-16 2982
-
锂离子电池的设计2013-05-20 3503
-
锂离子电池的性能2013-06-13 5631
-
锂离子电池的工作原理和使用注意事项2014-10-29 6438
-
锂离子电池和锂电池的区别2015-12-28 5808
-
锂离子电池的最新正极材料:掺锰铌酸锂?2016-01-19 6406
-
【转】锂离子电池的维护和保存技巧分享2016-08-18 4929
-
锂空气电池未来或击败锂离子电池2018-10-09 2323
-
锂离子电池简介2020-11-03 2859
-
锂离子电池的的原理、配方和工艺流程2021-04-07 4811
-
如何选择动力型锂离子电池的正极材料?2021-05-12 3311
-
磷酸铁锂——新型锂离子电池正极材料2009-10-27 1448
-
锂离子电池正极材料的发展趋势2009-11-09 966
-
磷酸铁锂锂离子电池正极材料2011-02-24 661
-
锂离子电池的工作原理与结构2021-03-10 18591
全部0条评论

快来发表一下你的评论吧 !
