

由无机ZnPS3固体电解质实现的稳定固态锌碘电池
电池技术
描述
研究背景
在当前发展的多种储能技术中,水系锌离子电池因其具有低成本、高功率密度及环境友好等优势而受到广泛关注。近年来,研究者们研究了多种锌离子电池体系,如Zn-Mn、Zn-V及Zn-I2等,其中,由于I2的储量丰富及理论比容量高,Zn-I2电池体系的发展尤为迅速。然而,长期以来,碘正极中的活性碘物种在电化学循环过程中存在溶解及穿梭的问题,易造成Zn-I2电池的不可逆容量损失及锌负极的不可逆腐蚀,进而加速电池的失效。针对这些问题,研究人员通常采用不同的策略,如设计多孔载体来吸附I2,加入中间层抑制多碘离子的穿梭,及利用催化剂来加速碘物种的氧化还原反应动力学等。经过改性后,尽管Zn-I2电池性能在很大程度上得到了改善,但多碘化物的穿梭问题仍无法被完全解决,以致目前的Zn-I2电池的循环性能仍不能满足实用化的需求。
研究内容
基于此,厦门大学赵金保教授/杨阳副教授团队开发了一种可导Zn2+离子的无机固态电解质ZnPS3,并首次成功将其用于Zn-I2电池的组装。ZnPS3电解质具有丰富且有序的导Zn2+通道,且Zn2+在ZnPS3晶体结构中的扩散能垒较低(≈0.3 eV),使其具有较高的离子电导率(2.0×10-3 S cm-1,30℃)。通过实验结果证明,基于无机ZnPS3电解质的固态Zn-I2电池可在 0.1 A g-1下稳定循环 400 次;原位拉曼和原位XRD结果表明,多碘离子的穿梭效应受到显著抑制,及固态Zn-I2电池主要是基于I-/I0氧化还原对进行可逆的充放电反应;此外,结合实验数据和理论模拟结果,进一步发现ZnPS3电解质的固态Zn2+传导机制有利于抑制由活性水分子引起的Zn金属负极的界面副反应,同时,ZnPS3电解质还可调控Zn2+的沉积/剥离行为,使其达到均匀沉积/剥离的效果。最后,无机固态电解质ZnPS3的兼容性也在Zn-CuS电池体系中得到证明,说明ZnPS3电解质可作为未来开发无机固态锌离子电池的一种潜在无机固态电解质。
其成果以题为“Stable Solid-State Zinc–IodineBatteries Enabled by an Inorganic ZnPS3 Solid Electrolyte withInterconnected Zn2+ Migration Channels”发表在国际知名期刊Adv. Funct. Mater.上。本文第一作者为2023级博士研究生吕泽恒,通讯作者为赵金保教授和杨阳副教授,通讯单位为厦门大学化学化工学院。
研究亮点
通过真空封管反应,成功制备出纯相、高结晶度的ZnPS3材料;
通过与疏水性PTFE结合,大大提高了ZnPS3无机固态电解质的空气稳定性;
基于固态Zn2+传导机制,多碘离子的穿梭得到了显著抑制,使得固态Zn-I2电池能在较低倍率0.1 A g-1下稳定循环 400 次;
实验数据和理论模拟结果表明,ZnPS3电解质还可调控Zn2+的沉积/剥离行为,使其达到均匀沉积/剥离的效果
无机固态电解质ZnPS3的兼容性也在Zn-CuS电池体系中得到证明。
图文导读
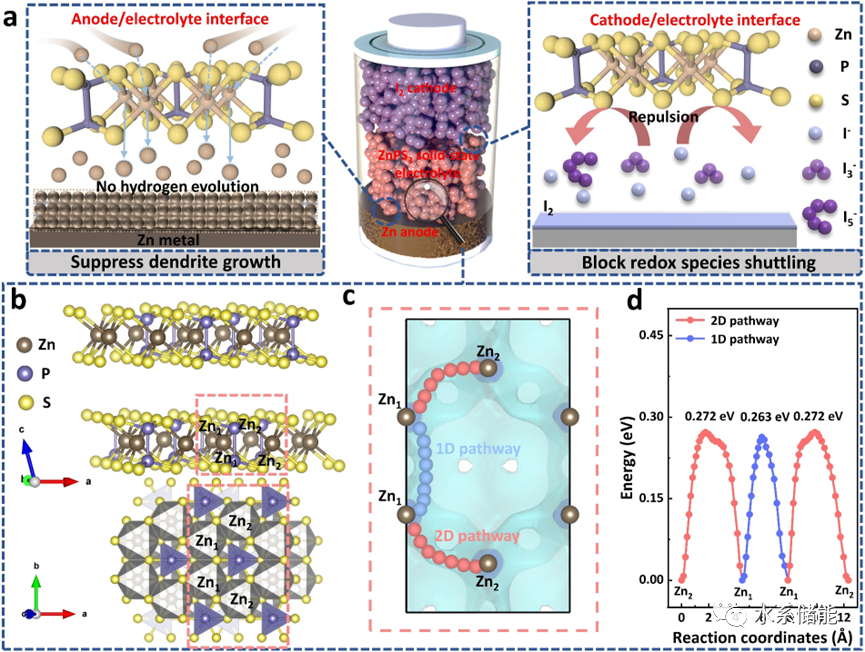
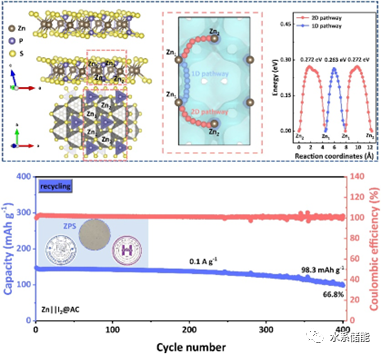
图1. a)提出的固态Zn-I2电池原型由Zn金属阳极、I2阴极和ZnPS3(ZPS)电解质组成。b)ZPS的晶体结构。c)ZPS晶体骨架中Zn2+迁移路径和d)相应的能垒。
▲由制备的ZnPS3无机电解质所组装的固态Zn-I2电池,其正极侧多碘化物的穿梭和Zn金属负极侧的析氢反应应均可得到有效抑制;通过BVSE理论计算,发现ZnPS3晶体结构中具有相互连通的多种Zn2+迁移路径,且整体的Zn2+迁移能垒约为0.3 eV。
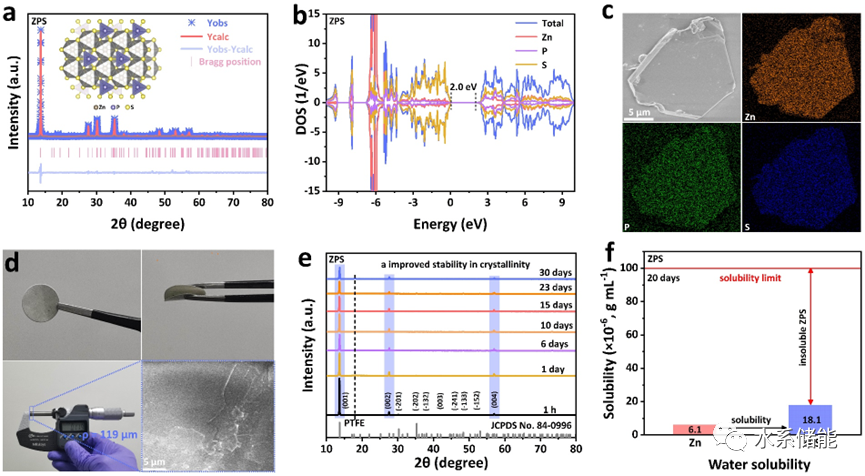
图2. a)ZnPS3(ZPS)的X射线衍射(XRD)图案及其Rietveld精修结果。b)ZPS的总态密度(DOS)。c)ZPS的扫描电子显微镜(SEM)图像和Zn、P、S元素分布图。d)ZPS电解质膜的光学和SEM图像。e)ZPS膜长时间浸入去离子水中的XRD结果。f)通过电感耦合等离子体光谱仪(ICP)分析将一块ZPS膜浸入去离子水中20天的结果来推测的ZPS的水溶性。
▲根据XRD表征及其精修结果,看出ZnPS3相纯且结晶度高;通过研究ZnPS3的电子结构,发现其能带间隙宽至2.0 eV,与电子绝缘的ZnS的带隙2.2 eV接近,说明ZnPS3电子绝缘的特性,这有利于其作为锌金属电池的电解质;通过扫描电镜观察,发现ZnPS3的二次颗粒为不规则的微米级别块状;通过与疏水性PTFE结合,ZPS电解质的空气稳定性得到大幅提升。
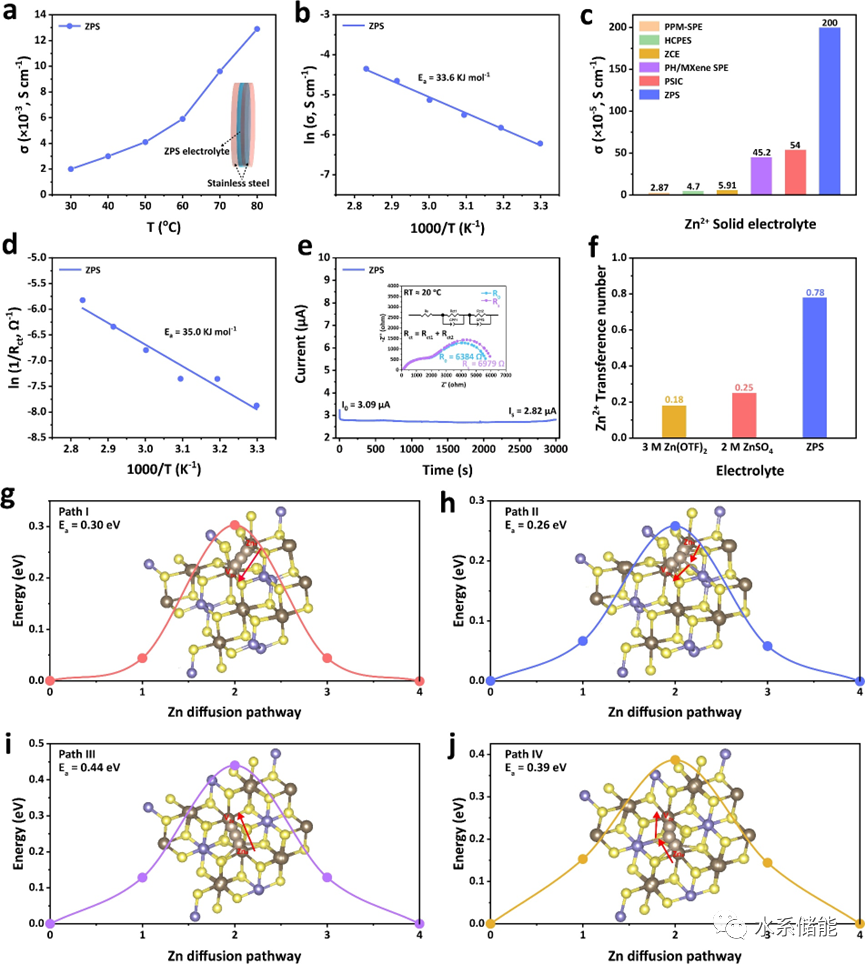
图3. a)ZnPS3(ZPS)从30℃到80℃的温度依赖性离子电导率。b)ZPS离子电导率的阿伦尼乌斯图。c)ZPS电解质与其他Zn2+固体聚合物电解质(SPE)的离子电导率比较。d)采用ZPS电解质的固态Zn||Zn对称电池中质量和电荷转移过程的总能垒。e)室温下使用ZPS电解质的Zn||Zn在恒电位极化(ΔV=20mV)下的电流变化,插图显示了极化前后的EIS。f)ZPS和液体电解质之间Zn2+迁移数的比较。g-j)四种可能的Zn2+结晶ZPS宿主中具有空位跳跃机制的迁移路径。 ▲在30℃时,ZPS的离子电导率为2.0×10-3 S cm-1,这与常规的液态电解液相比,仅相差一个数量级,而与目前所报道的用于锌金属电池的聚合物电解质相比,该离子电导率则处于较高的水平;利用阿仑尼乌斯方程,计算出了ZnPS3体相中的Zn2+迁移能垒约为33.6 kJ mol-1,而Zn2+迁移能垒与弱溶剂化Zn2+在电极界面脱溶剂化能垒之和为35.0 kJ mol-1;利用恒压极化测试,进一步测试了固态电解质的Zn2+迁移数为0.78,高于3 M Zn(OTF)2和2 M ZnSO4;最后,从理论模拟出发,模拟了Zn2+在ZnPS3体相中的四条迁移路径,计算所得的迁移能垒在0.26-0.44 eV。
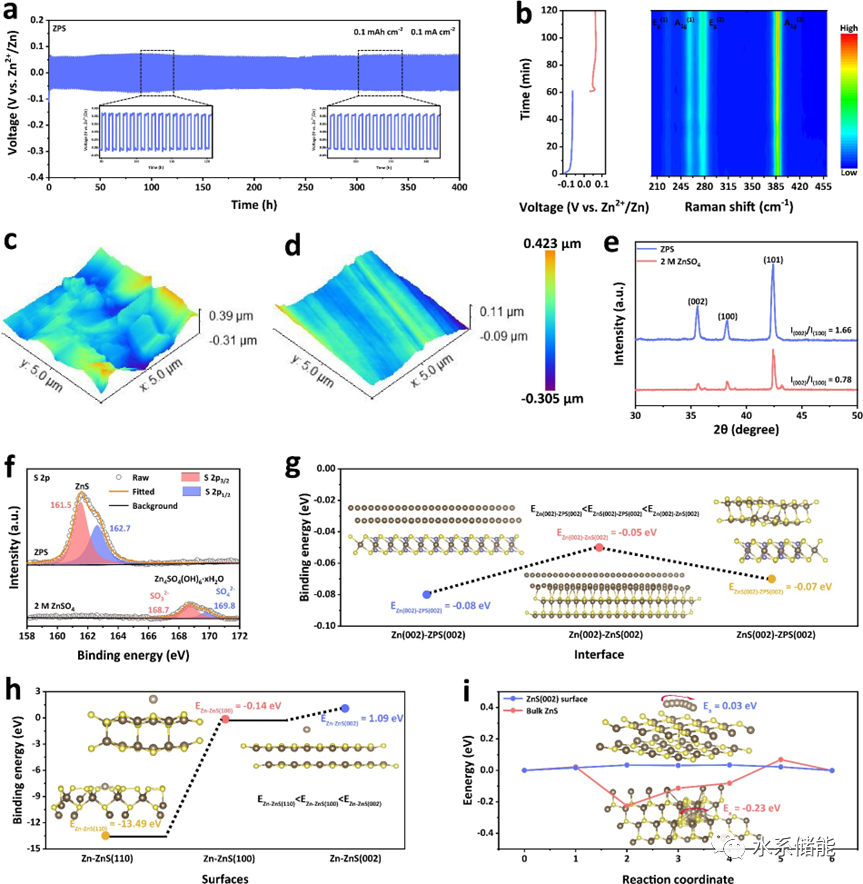
图4. a)在Zn||Zn对称电池中使用ZnPS3(ZPS)电解质在0.1 mA cm-2下以0.1 mAh cm-2容量进行恒电流Zn2+电镀/剥离。b)初始循环时使用ZPS电解质的Zn||Zn的原位拉曼结果。Zn电极在c)2 M ZnSO4和d)ZPS电解质中循环10次后的原子力显微镜(AFM)图像。e)使用2 M ZnSO4和ZPS电解质在Zn箔上电镀Zn2+10小时的掠入射X射线衍射(GIXRD)图案。f)Zn电极在2 M ZnSO4和ZPS电解质中循环五次后的S 2p X射线光电子能谱(XPS)谱。g)计算出的Zn(002)-ZPS(002)、Zn(002)-ZnS(002)和ZnS(002)-ZPS(002)的界面能。h)Zn-ZnS(110)、Zn-ZnS(100)和Zn-ZnS(002)的结合能。i)Zn2+沿ZnS(002)表面和块体ZnS扩散的势垒。
▲通过Zn||Zn对称电池的循环测试,可以发现,ZnPS3可支持Zn金属进行稳定的电化学循环;通过原位拉曼、AFM和掠入射XRD表征,发现ZnPS3在电化学循环过程保持结构稳定,且有助于Zn金属实现(002)晶面的优势沉积,得到较为平整的沉积形貌。进一步地,我们根据以往硫化物固态电解质在锂金属电池的研究,猜测在Zn金属与ZnPS3接触的界面处会形成一层ZnS层,且该ZnS层可能可以诱导Zn2+离子沿着(002)晶面方向进行沉积。通过理论计算,确实发现当在Zn金属与ZnPS3接触的界面处形成ZnS层时,该多相界面的兼容性会比两相界面更高,且Zn2+也会更趋向于沿着(002)方向进行沉积。
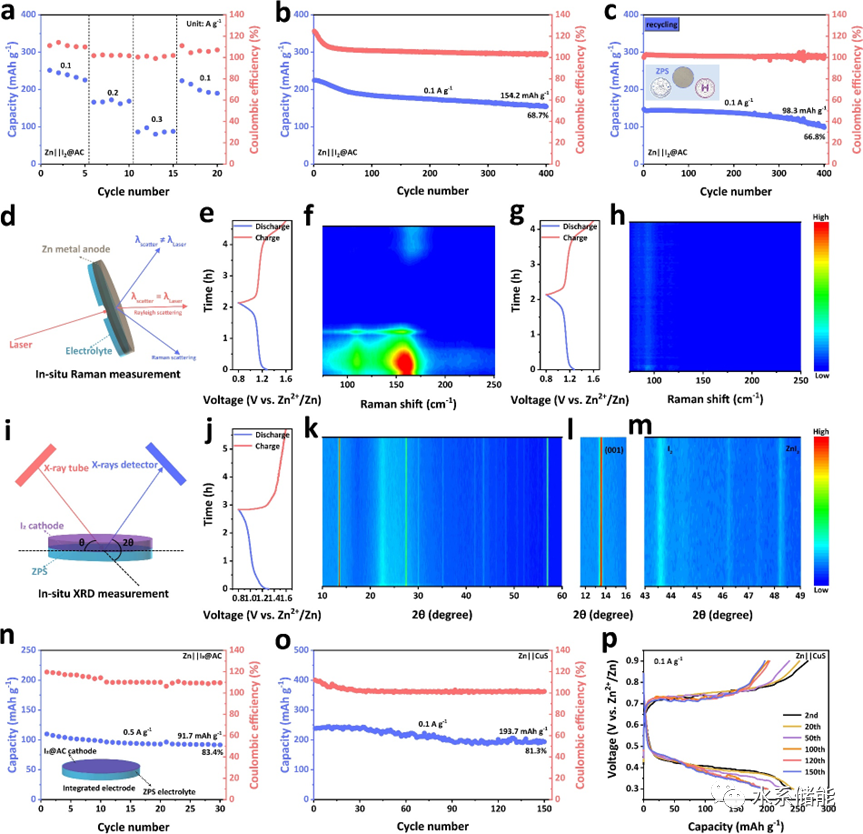
图5. a)Zn||I2@AC的倍率性能。b)Zn||I2@AC与新鲜 ZnPS3(ZPS)电解质和c)回收的Zn||I2@AC与可回收ZPS的循环性能。d)原位拉曼和i)原位X射线衍射(XRD)测量的简单示意图。f)2M ZnSO4和h)ZPS电解质中Zn||I2@AC的原位拉曼结果,以及e,g)相应的初始放电-充电曲线。j)使用ZPS电解质的Zn||I2@AC的初始放电/充电曲线和k-m)原位XRD测量的相应结果。n)使用集成电极的固态Zn||I2@AC的高倍率性能。o)使用ZPS电解质的Zn||CuS在0.1 A g-1下的循环稳定性和p)相应的放电-充电曲线。
▲通过与I2正极和CuS正极匹配,确定ZnPS3无机固态电解质可以成功应用于Zn金属电池的稳定循环。当对Zn||I2@AC体系进行原位XRD表征时,不仅发现ZnPS3的结构始终保持稳定,同时也检测到ZnI2的信号,说明I2正极在发生转化反应,更确切地证明了ZnPS3无机固态电解质可以传导Zn2+离子,且可以实现无机固态Zn金属电池的组装与循环。
研究总结
在这项工作中,研究人员采用具有高离子电导率、低Zn2+离子迁移能垒和优异的化学/电化学稳定性的无机固态ZPS电解质成功组装了固态Zn-I2电池。ZPS的高Zn2+迁移数和非去溶剂化固态Zn2+离子导电机制,抑制了Zn金属阳极的枝晶生长,达到了优先Zn(002)沉积取向的效果。原位拉曼和XRD结果进一步证实了ZPS电解质可有效阻挡多碘化物穿梭,确保了基于I-/I0氧化还原对的转化反应的可逆进行。最后,Zn||I2@AC和Zn||Cu全电池的成功应用证明了ZPS电解质可作为一种有前途的无机固态Zn2+离子导体,为使用无机晶体电解质开发实用的固态锌基电池开辟了新的可能性。
审核编辑:刘清
-
固体电解质的物理性质如何?2019-09-17 2652
-
新型锌碘电池获新进展 将更安全更环保2018-09-19 4132
-
基于溶液制造固态电池电解质2020-03-23 3026
-
剖析稳定锂金属电池的长效固体电解质界面2021-06-04 3681
-
钠离子电池的电解质分类2022-10-09 6779
-
改变电解质分布调控固态界面实现高性能固态电池2022-10-21 4194
-
固态电池电解质的分类及性能对比2022-11-30 20749
-
聚合物电解质离子电导率及界面稳定性的影响因素2023-02-03 5886
-
高电压稳定的固态电解质实现高能量、高安全的固态锂金属电池2023-03-27 2399
-
钠-钾电解质界面相实现室温/0°C固态钠金属电池研究2023-03-30 1878
-
固态电解质电导性 (Solid系列)2023-06-25 2314
-
一种有机-无机非对称固态电解质,实现长循环稳定的高压锂电池2023-12-10 4593
-
固态电池的概念_固态电池的发展趋势2024-09-15 9822
-
Li3MX6全固态锂离子电池固体电解质材料2025-01-02 2538
-
氨基磺酸固态电池电解质2026-06-22 35
全部0条评论

快来发表一下你的评论吧 !
