

材料科学迈向AI4Materials的关键因素(上)
描述
摘要
材料的结构直接决定了其物理和化学性质,这种关系也常称之为“构效关系”。如何快速有效地建立起这种“构效关系”是探索和设计新型功能材料的关键。理论模拟能够根据材料的原子级构型较为精确地计算出材料的各种性质,对实验具有指导意义,然而对复杂体系的模拟计算过去通常受限于计算机的性能和成本,尤其是精确的密度泛函理论模拟计算。通用计算显卡(General-Purpose Computing on Graphics Processing Units,GPGPU)的发展可改善这种情况,在GPGPU加速的情况下,材料模拟所需的时间将会显著缩短。近年来,人工智能在各个领域展现出令人瞩目的能力,其中GPGPU扮演着至关重要的角色。
人工智能与材料科学在GPGPU的助力下有机地结合在一起,使得材料科学的研究模式进入一个新的阶段:AI4Materials,有望大幅加速新材料的研发。接下来,让我们一起来了解材料科学迈向AI4Materials的关键因素。本文将从密度泛函理论出发,首先介绍其理论框架的发展,其次讨论基于密度泛函理论的常用软件及其特点,同时介绍GPGPU对这些软件的加速效果,最后简要介绍材料基因组计划。下篇将介绍AI4Materials中的“材料大数据”以及人工智能算法结合材料科学取得的一些进展。
01
材料科学研究范式4.0:AI4materials
每种材料都是由众多原子按特定方式排列组合而成的,排列方式决定了材料的物理和化学性质,这种结构与性质之间的关系常被称为“构效关系”。寻找并理解这种关系,以及根据它来设计新材料,一直是科学家们不断努力的方向。
传统的新材料开发通常采用试错法,然而其实验步骤繁琐、周期长且成本高,制约了材料研发的速度。随着理论的发展和计算机技术的进步,理论模拟计算已成为发现新材料的重要方法之一。如基于密度泛函理论的电子结构计算、蒙特卡罗模拟、分子动力学模拟、相场法和有限元分析等高精度计算方法被广泛应用于新材料的探索和发现。然而,这些方法通常仅适用于特定系统,对于复杂系统的计算则具有一定挑战性。近年来,GPGPU的快速发展正在改善这一情况。此外,目前的理论发展仍不足以满足对材料定量表征的需求。因此,亟需开发新的方法和工具来指导新材料的探索和设计。
随着实验研究和材料理论的发展,上述实验和计算模拟产生了大量数据。在材料科学研究范式经历了实验科学、理论科学和模拟计算三个阶段后,长期积累所形成的“材料大数据”为材料科学步入“第四范式”提供了“养料”,如图 1所示,大数据和人工智能的结合被称为“科学的第四范式”或“第四次工业革命”。机器学习已成为材料信息学的核心技术之一,以材料数据库为基础的机器学习能够快速实现材料的预测,有望加快新材料的设计,缩短材料的开发周期。即使在不了解潜在物理机制的情况下,机器学习也能够从现有数据中学习其蕴含的行为和趋势,在材料科学中正在发挥越来越重要的作用。
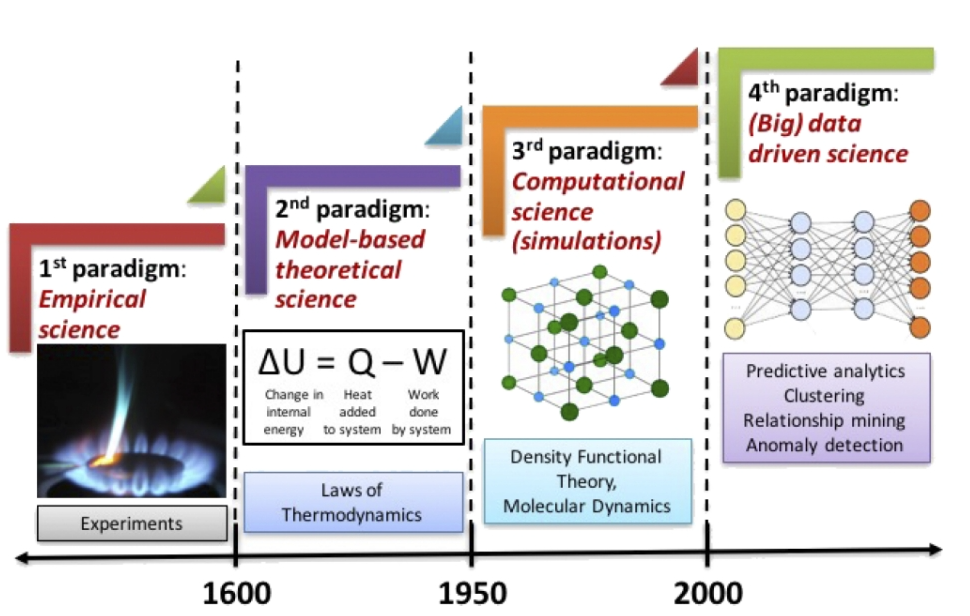
图 1 材料科学研究四个范式。
图片来源于文献[1]
02
密度泛函理论扮演的角色
密度泛函理论(Density Functional Theory, DFT)是量子力学中一种重要的计算方法,被广泛应用于研究原子、分子核凝聚态物质的性质。DFT可用于准确地描述材料和分子的电子结构,通过计算电子密度来获得分子结构、能量、电子亲和力、离子化势等物理和化学性质。借助 DFT,科学家可以设计新型材料并预测其性质,包括但不限于催化剂、半导体、超导体等,为实验室合成和材料应用提供理论指导。例如,DFT 能够揭示催化剂表面上的吸附、反应和表面结构,帮助解释催化作用的机理,并加速新型催化剂的设计与优化。在能源研究中,DFT可用于模拟电池材料的性能、储能材料的吸附特性以及光催化剂的能量转换过程。DFT还可用于理解生物分子(如蛋白质和酶)的结构和活性,帮助科学家解释生物化学过程和生物分子的相互作用。
二维材料通常指只具有几个原子厚度的平面薄膜材料,这一概念起源于2004年,当时曼切斯特大学(University of Manchester)Geim小组成功分离出单层原子层的石墨材料:石墨烯(Graphene)。由于Geim在实验中(通过机械剥离法)发现了二维石墨烯,他获得了2010年的诺贝尔物理学奖,这标志着一个新材料领域的诞生:二维材料。可以想象一下,二维材料的原子级厚度会限制电子的运动,使其呈现出与三维材料不同的性质,这种效应也称为量子限域效应。因此,单原子层厚度的石墨烯具有出色的特性,如高载流子迁移率、高开关比、高力学强度(在二维平面内比金刚石还要硬,当然了,在垂直平面的方向上是很软的)等等。目前,基于石墨烯的研究和应用已经取得了长足的进步,如图 2所示,并将对我们的生活产生深远影响。

图 2 石墨烯材料发展中的重要事件时间线[2]
除了石墨的单层结构有这种特性,其他层状材料的单层是否存在?除了碳(C)元素,其他元素的组合形成的二维材料也是否存在?性质如何呢?再进一步,二维材料是否可以在三维空间“搭积木”?这些都引起了科学家们极大的兴趣。如果理论上能够快速、准确模拟计算出材料的性质,例如稳定性、电子结构、带隙、电荷传输等。这种模拟方法可以为科学家提供宝贵的信息,具有指导意义,帮助筛选出具有特定性质的候选物质,以便进行进一步的实验验证和研究。这种筛选方法可以节约大量的时间、精力和资源,提高实验工作的效率。
经过科学家多年的努力和耕耘,已经发展出了很多方法来模拟计算材料的基础性质。在这些方法中,DFT由于能够较为精确地模拟计算出基态性质而被广泛应用。通过DFT模拟计算,我们可以轻松地获得石墨烯的基态性质。如图 3所示,根据密度泛函理论可以计算出石墨烯的电子能带图和声子谱,其结果与实验值符合得非常好。结合理论模拟计算和实验制备与测量,科学家们已经发现了多种二维材料,如二维氮化硼、二维硫属族化合物、MXene材料等,以及如何用这些层状二维材料“搭积木”(称为范德华异质结,van der Waals heterostructures。注:二维材料层与层之间靠van der Waals作用力连接。)来获得不同的物理、化学性质,如图 4所示。
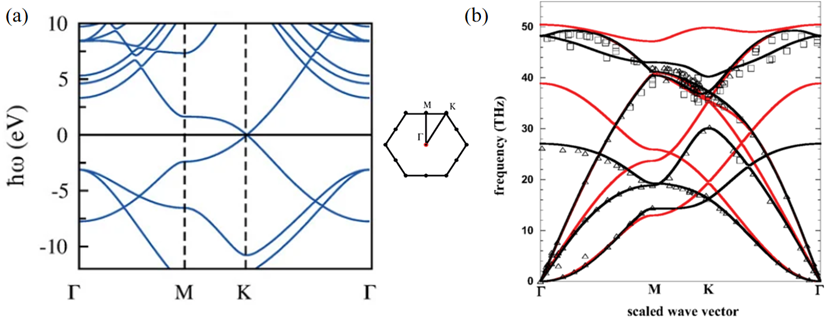
图 3 石墨烯的密度泛函理论计算结果。
(a)石墨烯的电子能带结构[3]。其中费米面设为0 eV。(b)石墨烯的声子谱[4]。其中,黑色实线是DFT计算值,空心三角形和方形散点是实验值,红色实线是基于优化的Tersoff经验势计算的结果。中间的六边形是石墨烯的K空间及高对称点

图 4 (a)二维材料所组成的“积木”(异质结)示意图。(b)典型的二维材料家族。
图片来源于文献[5]。
近年来,“魔角”石墨烯的发现引领了转角电子学领域的发展[6–9]。在小角度转角(约1.08°)下,双层石墨烯表现出在费米面附近出现平带的电子结构,这导致其显示出神奇的超导效应。“魔角”石墨烯的晶胞包含了11164个原子,每层石墨烯含有5582个碳原子。密度泛函理论的模拟计算也证实了魔角石墨烯在费米面附近出现平台的电子特征[10],如图 5所示,然而,计算这一电子结构需要使用2880至5760个CPU核计算约30天。这种耗时的模拟计算在一定程度上限制了科学家们对大体系新材料的研究和探索。随着GPGPU的发展,其强大的计算能力将大大加速科学家们对大体系和新材料的研究进展。以双层石墨烯转角5.086°(包含508个C原子)为例,在GPGPU上利用密度泛函理论计算其电子能带结构仅需要4.1个小时,如图 5所示。随着GPGPU的快速发展,基于密度泛函理论进行高精度大体系的模拟计算将会变得“唾手可得”。
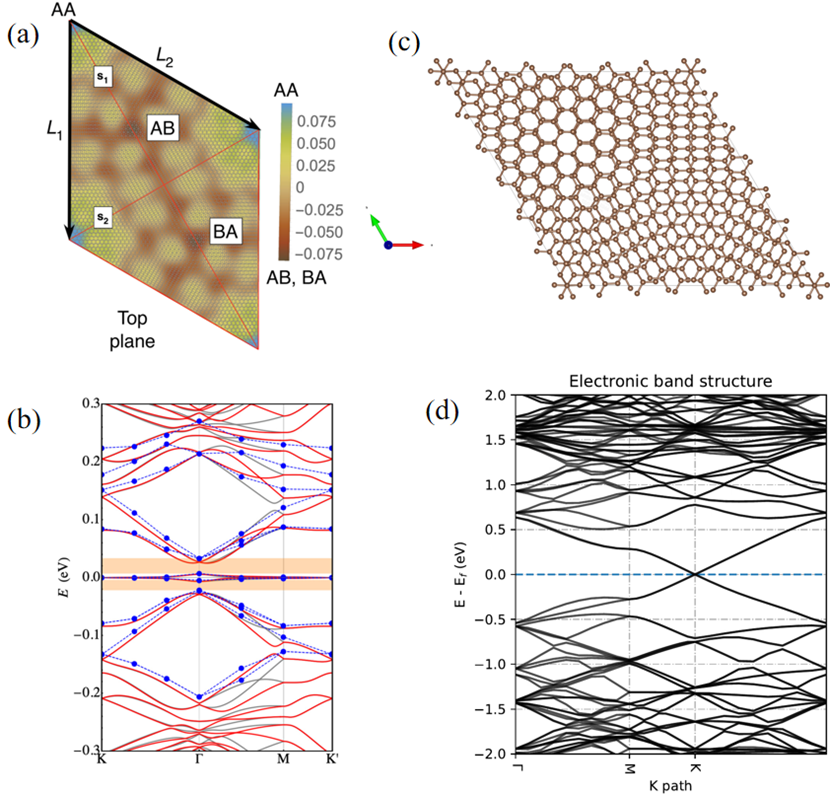
图 5 转角石墨烯结构与基于密度泛函理论模拟计算的电子能带图。(a)转角为1.08°的魔角石墨烯原胞,包含11164个C原子,其中颜色表示层间不同的原子间距[10]。(b)在2880至5760个CPU核计算出的基于密度泛函理论的电子结构[10],其中费米能设为0 eV。(c)和(d)是转角为5.086°双层石墨烯在GPGPU上的基于密度泛函理论计算出的结果。(c)经过结构弛豫后的原胞,其中包含508个C原子。(b)电子能带结构,其中蓝色虚线是费米能级位置(0 eV)。
通过上面的介绍,我们可以看出密度泛函理论的计算结果与实验测量非常吻合,已成为材料性质预测和物理理论研究中的重要工具,对实验研究具有一定的理论指导意义。然而,高精度密度泛函理论模拟大体系十分昂贵且耗时,这限制了其应用范围。但随着GPGPU技术的发展,密度泛函理论的计算效率将会大大提高。
为什么密度泛函理论能够准确地计算材料的基态性质呢?接下来,让我们回顾一下密度泛函理论的发展历程,了解科学家们在这一领域的贡献,感受物理学家智慧的闪光点。在下一节的介绍中,我们将探究这一理论的发现和演变,以及背后的科学思想。
03
密度泛函理论发展历史
在海森堡、薛定谔和狄拉克等人相继建立非相对论和相对论的量子力学后,狄拉克在1929年提出:“The fundamental laws necessary for the mathematical treatment of large parts of physics and the whole of chemistry are thus fully known, and the difficulty lies only in the fact that application of these laws leads to equations that are too complex to be solved.” (处理大部分物理学和整个化学所需的数学基本定律已完全了解,困难仅在于应用这些定律会产生过于复杂而无法解决的方程。)[11]。以当时的计算能力,想通过求解量子力学方程来严格地进行材料的理论计算是根本不可能的。自1960年代初以来,经过众多物理学家、数学家和化学家的努力,特别是计算机技术的进步,这种情况得到了很大的改变。目前,基于密度泛函理论的第一性原理计算已经成为科学研究的重要方法之一。更值得一提的是,在GPGPU算力的支持下,密度泛函理论的模拟计算速度得到了显著提升。
密度泛函理论由于其清晰的物理原理,相对较小的计算量以及较为准确的计算结果,在当前广泛应用于材料性质的理论模拟计算。密度泛函理论的发展过程体现了物理学家们的努力和智慧。
如果一个函数的变量也是一个函数,则称为泛函,它是函数的函数,也常称“函数的函数就是泛函”。通常,泛函是一个积分,这是因为积分表示了函数整体的特点。如果只是涉及函数在某一位置的信息,如F=exp(cos(θ)),它可以说是cos(θ)的函数,但只是在θ处计算,因而是一般的函数,不必归之于泛函。对于密度泛函理论,其表述的是材料基态能量是电子密度的泛函(基态能量是电子密度的积分),而电子密度又是位置坐标的函数。
1927年,Thomas和Fermi基于理想状态下的均匀自由电子气体模型,认为能量是电子密度的泛函,其中,电子密度是位置的函数。该模型包含了体系的基态能量,考虑了动能项以及核与电子、电子与电子之间的库伦相互作用。然而,Thomas-Fermi模型并未考虑电子之间的交换关联作用,后来,Dirac于1930年对这个方法进行了扩展,把局部近似的电子间交换作用也表示成电子密度泛函的形式,并作为Thomas和Fermi模型的补充,即Thomas-Fermi-Dirac模型,此模型相比较薛定谔方程有了非常大的简化,是密度泛函理论的雏形,但是由于其近似过于粗糙,计算出的结果误差比较大。
到了1963年,Walter Kohn在巴黎高等师范学院(École Normale Supérieure)阅读一些关于冶金相关的文献时发现,合金中原子的有效电荷的概念非常有趣,它能粗略地表征原子间的电荷转移,这里的有效电荷在坐标空间是局域化的,这种观点与在动量空间中的非局域化波是不同的,如周期性边界条件下的Bloch波。Walter Kohn联想到Thomas-Fermi近似中能量可以表示为电子密度的泛函,并敏锐地发现,对于单个粒子,势能V(r)和基态的电子密度n(r)之间存在明确的基本关系,那么对于包含N个电子系统,其电子密度也决定了这个体系的总哈密顿量H。
接着,Walter Kohn用他“擅长”(“beloved”)的Rayleigh Ritz变分原理仅用三行论证就证实了这个猜想,以至于当时他都不敢相信自己这么轻易就得到了这个如此了不起的结果[12]。接着,Walter Kohn邀请Pierre Hohenberg加入进来,首先是调研文献,看这个简单的结果是否已知(显然是没有),并短时间内,根据电子密度n(r)而不是多电子波函数得到基态能量的Rayleigh-Ritz定理,也就是现在的Hohenberg Kohn (HK)变分原理,并于1964年发表[13]。尽管这个工作并没有给出泛函的具体形式,但是毋庸置疑的是,HK定理直接奠定了密度泛函理论的理论根基。不久之后,Walter Kohn回到了加州大学圣地亚哥分校,和刚来的博士后Lu Jeu Sham一起从HK定理出发,推导出了现在被称为Kohn-Sham方程的结果,即下式,并于1965年发表[14]。
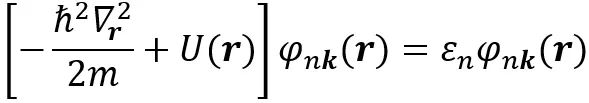
其中,中括号第一项是动能项,第二项是等效势能项,φnk(r)是波函数,n是能带指标,k是倒空间K点位置,r是实空间位置。
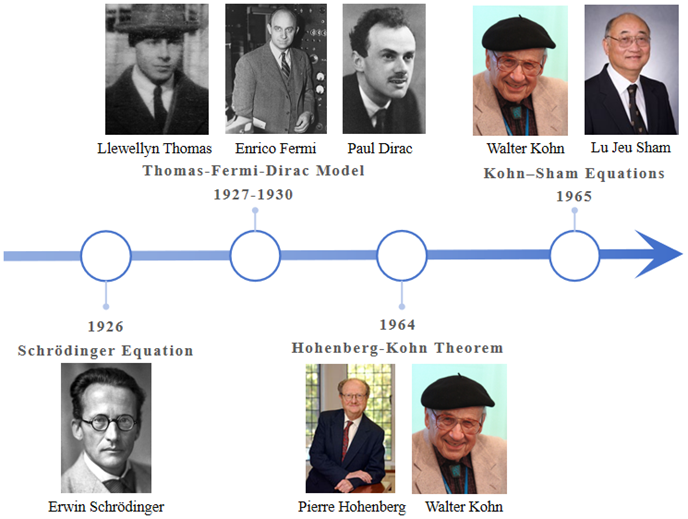
图 6 密度泛函理论发展历史
总的来说,密度泛函理论的发展历史大致可分为三个阶段,如图 6所示:
第一阶段
Thomas和Fermi基于理想状态下的均匀自由气体模型,把电子密度作为能量的泛函,首次引入密度泛函的概念,但是处理太过粗糙;
第二阶段
Pierre Hohenberg和Walter Kohn用简洁的方式证明了两个重要的定理,为密度泛函理论奠定了理论基础,但没有给出泛函的具体表达式;
第三阶段
Walter Kohn和Lu Jeu Sham提出了非相对论密度泛函理论的具体实现方法。
受到Hatree-Fork近似的启发,Kohn和Sham用一个非相互作用的电子系统代替具有相互作用的电子系统,把由具有非相互作用电子近似带来的动能和势能的误差放在交换关联项中。自从Kohn-Sham方程提出以来,DFT方法一直是物理、化学领域计算电子结构及其特征最有力的工具之一。因此,Walter Kohn因提出DFT方法获得1998年诺贝尔化学奖(与之分享的是John A. Pople),表明DFT方法在量子化学计算领域的核心地位和广泛应用。在1964年的时候,开发高效的计算机代码是物理理论学家面临的主要任务之一,其中John A. Pople为整个量子化学方法论的发展做出了巨大贡献。Pople将计算化学理论方法工具化,开发了Gaussian计算机程序,并于1970年发布第一个版本,而且多年来不断更新发展。目前Gaussian程序已经成为许多高校、研究所和商业公司重要的研究工具。

图 7. 1998年诺贝尔化学奖由沃尔特·科恩(Walter Kohn)和约翰·A·波普尔(John A. Pople)平分,以分别表彰他们对密度泛函理论和量子化学计算方法发展的贡献。
04
典型的基于密度泛函理论的材料模拟软件
基于密度泛函理论的第一性原理有很多软件,这里简单介绍一些典型和常用的软件平台,如The Vienna Ab initio Simulation Packag(VASP)、CP2K、Gaussian、Materials Studio(MS)、Quantum Espresso(QE)等软件。

图 8 一些典型的第一性原理密度泛函理论计算软件平台
The Vienna Ab initio Simulation Packag(VASP)是维也纳大学Hafner小组开发的基于密度泛函理论的材料模拟计算软件包,能够进行电子结构计算和量子力学-分子动力学模拟,由于其简单易用,准确性好,是材料模拟和计算物质科学研究中最流行的商用软件之一。VASP是在密度泛函理论(DFT)框架下计算多体Schrödinger方程的近似解,求解Kohn-Sham方程,或者在Hartree-Fock(HF)近似下解决Roothaan方程。VASP还实现了将Hartree-Fock方法与密度泛函理论混合的混合泛函。此外,还提供了Green函数方法(GW准粒子和ACFDT-RPA)和多体微扰理论(二阶Møller-Plesset)。在VASP中,一些核心量都是由以平面波基组来表示的,如单电子轨道、电子电荷密度和局域势。电子和离子之间的相互作用使用模守恒赝势、超软赝势或投影缀加波方法进行描述。为了确定电子基态,VASP利用了高效的迭代矩阵对角化技术,如迭代子空间下的残差最小化方法(RMM-DIIS)或Blocked Davison算法。这些与高效的Broyden和Pulay密度混合方案相结合,以加快自洽循环速度。
在VASP(5.4.4)中基于MPI的并行过程如图 9所示,在计算KS轨道的过程中,可以通过KPAR,即倒空间K点个数,来控制同时计算K点个数,以及NCORE来控制同时计算每个K点上的能带个数。这种并行方式,尤其K点可以独立计算的这种特性也非常适用于GPGPU平台的加速计算。在这种并行框架下,VASP提供了基于CUDA和OpenACC的GPU加速方案,并在VASP.6.2.0之后全面转向OpenACC加速方案。在OpenACC加速方案下,VASP6.1.0版本(2020年)在GPGPU下的计算速度约是纯CPU的2.6-5.1倍[15],经过多次版本的更新以及GPGPU的发展,VASP6.4.1版本(2023年)在GPGPU下的计算速度约能达到纯CPU的8.2倍[16]。另外,从VASP.6.3.0版本开始,还支持on-the-fly机器学习力场,这进一步加快整个计算过程。
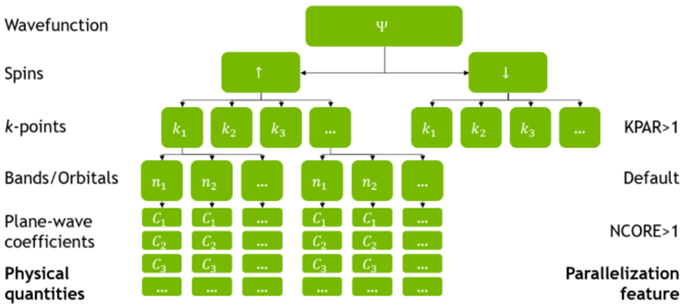
图 9 VASP 5.4.4的并行框架[17]。在求解KS波函数(Wavefunction)过程中,KPAR控制K点的并行度,NCORE控制Bands/Orbitals的并行度
CP2K是开源的用于量子化学和固体物理的第一性原理材料计算和模拟软件,可以模拟固态、液态、分子等材料体系。CP2K使用Fortran2008编写,可以利用多线程、MPI和CUDA的组合高效地并行运行,能够轻松模拟计算包含上千原子的体系。CP2K最开始是由马克斯-普朗克研究中心于2000年发起的一项用于固体物理研究的项目,现在已转由苏黎世联邦理工学院和苏黎世大学维护,成为一个开源的项目,遵从GPL协议。相比较VASP,CP2K输入文件的结构比较复杂,没有像VASP wiki类似的比较完整的使用和学习文档,学习曲线较为陡峭;对导体、磁性和小体系计算不太合适;但是CP2K中基于第一性原理的分子动力学模拟功能非常优秀。CP2K也开发了GPU相关的加速方案,基于GPGPU的计算相比较于纯CPU计算约能达到3.7倍的加速效果[18]。
Gaussian是一个量子化学软件包,它是目前应用最广泛的计算化学软件之一。如前所述,其代码最初由理论化学家、1998年诺贝尔化学奖得主John A. Pople编写,其名称来自于Pople在软件中所使用的高斯型基组,使用高斯型基组是Pople为简化计算过程缩短计算时间所引入的一项重要近似。其前期是免费的,后期发展成为成熟的商用软件,目前已经发展到Gaussian 16,可用于预测化合物和反应在各种化学环境中的能量、分子结构、振动频率和分子性质。Gaussian 16的模型既可以应用于稳定物质,也可以应用于难以或不可能通过实验观察到的化合物,无论是由于它们的性质(例如,毒性、可燃性、放射性)还是它们固有的转瞬即逝的性质(例如,短时间内的中间体和过渡结构)。而且,Gaussian 16提供了用户友好的界面(GaussianView),大大降低了科研工作者从事量子力学计算模拟的门槛。并且,Gaussian 16支持GPU的加速方案,在8张GPGPU上的加速效果约是纯CPU的2.3-4.2倍[19]。
BIOVIA Materials Studio(MS)是一个商业软件,包含完整的建模和仿真环境,旨在使材料科学和化学研究人员能够预测和理解材料的原子和分子结构与其特性和行为的关系。使用MS,许多行业的研究人员正在设计各种类型的性能更好的材料,包括催化剂、聚合物、复合材料、金属、合金、制药、电池等。Cambridge Sequential Total Energy Package(CASTEP)是MS软件中的第一性原理计算模块,用Fortran 2003语言编写,其基本可实现VASP中的主要功能,与VASP不同的是其在MS上提供了可视化、用户十分友好的操作界面。值得一提的是,CASTEP的Linux学术版是免费的。CASTEP的OpenACC加速方案还在积极开发中,在GPGPU的加速下,其计算速度约能达到纯CPU的2倍[20]。
Quantum Espresso(QE)(前期被称为PWscf)是一款开源的、被广泛使用的第一性原理密度泛函理论计算软件。与VASP一样也是基于平面波基组和赝势,相比较于VASP,QE具有开源免费、功能更加丰富、自带后处理程序等特点。本着开源项目的精神,QE已经发展成为一个独立且可互操作的代码分发版。QE发行版由一组“历史”核心组件和一组执行更高级任务的插件组成,以及许多旨在与核心组件互操作的第三方软件包。鼓励活跃在电子结构计算领域的研究人员通过贡献自己的代码或将自己的想法实施到现有代码中来参与该项目。自QE v.6.4版本后,QE开始加入了基于CUDA的GPU加速方案,QE-GPU v.6.5(2020年)在GPGPU上的加速效果是纯CPU的1.4-4.6倍[21]。
还有很多优秀的第一性原理计算软件,例如WIEN2K[22]、ORCA[23]、SIESTA[24]、ABINIT[25]以及国产软件RESCU[26]、ABACUS[27]等,这里就不一一介绍了。
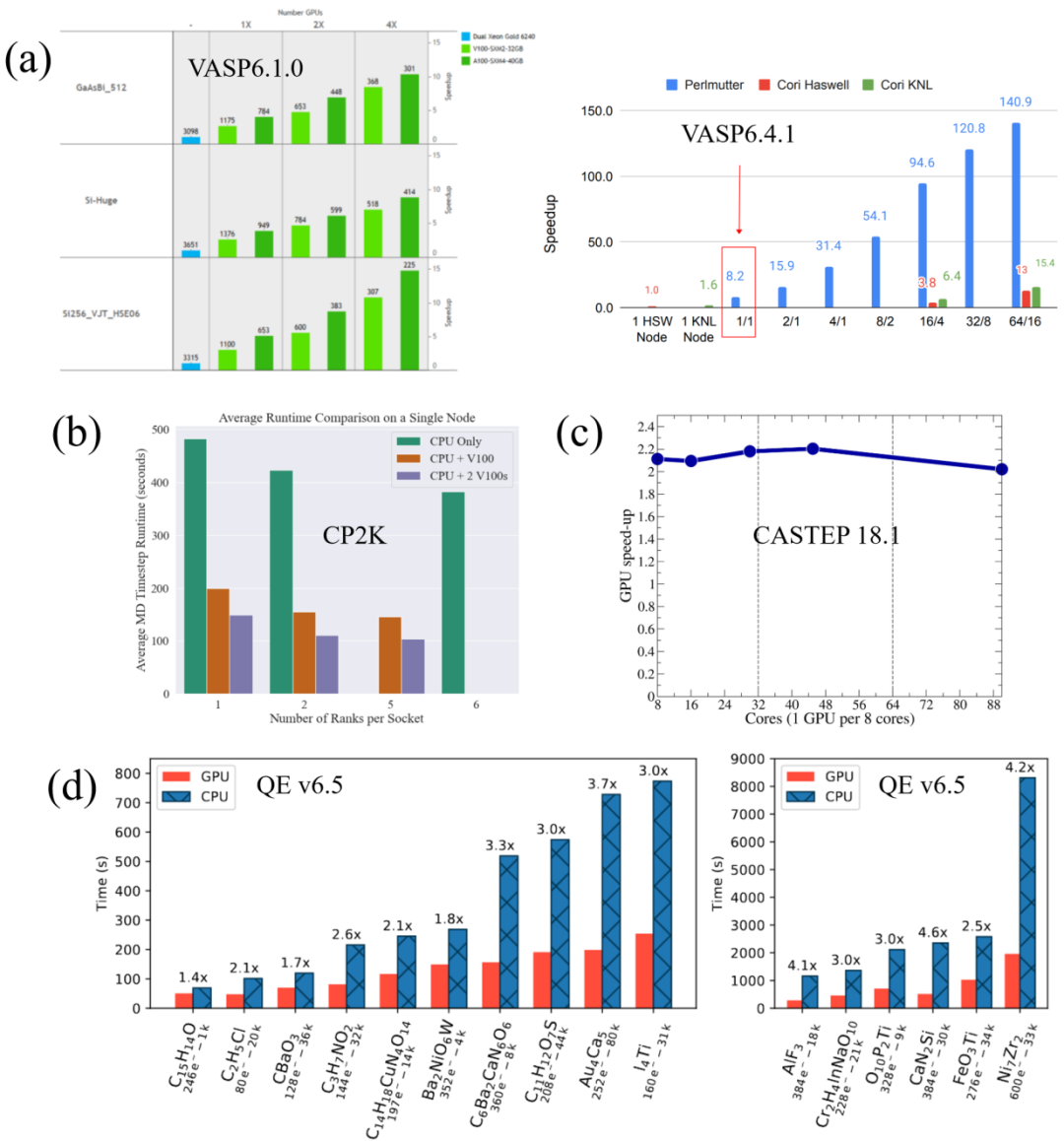
图 10 在不同密度泛函理论模拟计算软件中GPU相对于CPU的加速效果。(a)VASP,(b)CP2K,(c)CASTEP,(d)QE
图片来源于文献[15,16,18,20-21]。
05
材料基因组计划
新材料是人类文明社会进步的基石,新材料的革新对技术的进步和产业的发展发挥着重要的作用。材料基因组计划(Materials Genome Initiative,MGI)于2011年6月24日由美国率先提出,如图 11所示,其是从应用需求出发,倒推符合相应结构功能的材料,旨在通过新材料研制周期内各个阶段的团队相互协作,加强“官产学研用”相结合,注重实验技术、计算技术和数据库之间的协作和共享,达到缩短新材料研发周期的目的,降低研发成本。材料基因组计划的一个突出重点是强调材料计算模拟、实验和数据库的协同性对加快材料研发的贡献。
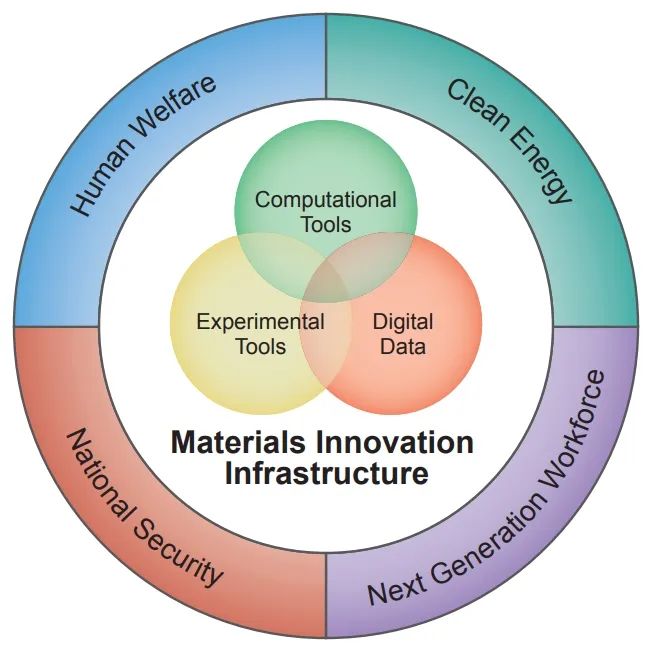
图 11 材料基因组计划(Materials Genome Initiative,MGI)[28]
中国也重点专项全方位布局研究材料基因组计划。材料高效计算、高通量实验和大数据技术构成材料基因工程的基础技术体系。通过材料高效计算和高通量实验,可实现新材料的快速筛选和材料数据的快速积累;通过大数据和人工智能技术的应用,可实现材料成分和工艺的全局优化、材料性能的提升;通过创新平台,实现材料基因工程关键技术的深度融合和协同创新。材料基因工程关键技术的应用,将材料传统顺序迭代的试错法研发模式,变革成全过程关联并行的新模式如图 12所示,全面加速材料发现、开发、生产、应用等全过程的进程,促进新材料研发和工程化应用[29]。
通过MGI项目,材料科学家们可获得更多的工具和资源,可更快、更高效地进行新材料的研发。虽然短期内具体新材料的商业应用可能有限,但长期来看,MGI的努力有望加速材料创新的进程,推动科学、工业和社会的发展。
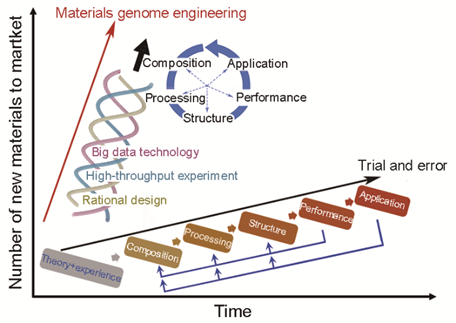
图 12 材料基因组工程变革研发模式。
图片来源于文献[29]
06
小结
通过上面的介绍,我们可以了解到在材料研究的第三和第四范式中,密度泛函理论占据着重要的地位。密度泛函理论作为第一性原理计算的基础工具,为研究人员提供了解释和预测材料性质的强有力手段。它不仅推动了新型功能材料的探索,还为实验研究提供了理论指导,并为材料研究的第四范式提供了可靠的数据支持。尽管密度泛函理论在几十年的发展中取得了长足进步,但随着研究体系的增大和精度要求的提高,计算机算力一直是限制其应用的主要因素。然而,随着通用计算(GPGPU)技术的发展,其为材料模拟计算提供了强大的算力支持,使得理论上精确模拟计算大体系基态性质成为可能。随着社会对新型功能材料需求的不断增加,传统的研究模式已经不能满足需求。因此,材料基因组计划的出现对加速新材料开发和应用具有重要意义,其中,GPGPU提供的高算力也扮演着关键的角色。这一计划将指导并加速新型功能材料的发现和应用,为社会发展提供更多可能性。
07
预告
随着材料基因组计划的提出,在科学计算和人工智能算法的不断发展下,AI4Materials正在如火如荼地进行着,材料科学研究模式正在发生前所未有的变革,展现出巨大的潜力。接下来(下篇),我们将介绍不断发展的晶体材料数据库、科学计算对材料模拟计算加速以及人工智能算法在材料科学的应用。
审核编辑:刘清
-
电子背散射衍射技术(EBSD)在材料科学中的应用与解读2024-12-16 1880
-
名单公布!【书籍评测活动NO.44】AI for Science:人工智能驱动科学创新2024-09-09 3516
-
安泰高压放大器在极化特性及在材料科学中的应用2024-09-04 1240
-
科研进阶!EPR在材料科学中的应用交流会即将开幕2023-11-21 1267
-
中科曙光异构智能算力技术加速计算材料科学创新变革2023-08-01 1916
-
热像仪助力材料科学与工程研究(四)2022-08-18 1437
-
热像仪助力材料科学与工程研究(三)2022-08-16 2205
-
热像仪助力材料科学与工程研究(二)2022-08-02 1696
-
热像仪助力材料科学与工程研究(一)2022-07-22 1729
-
材料科学在医疗器械研发中的应用2020-04-14 6840
-
计算机在材料科学中的应用详细中文资料概述(免费下载)2018-06-05 1551
-
材料科学基础课件免费下载田民波老师2008-09-23 1773
全部0条评论

快来发表一下你的评论吧 !
