

利用太阳辐射直接修复富锂富锰正极!
描述
研究背景
富锂(Li)和富锰(Mn)层状氧化物材料(LMRO)因其高能量密度而被认为是最有前途的下一代电池正极材料之一。然而,循环过程中晶格氧的氧化还原导致局部缺陷的形成,以表面气体释放的形式出现氧损失,以及过度金属(TMs)的迁移,导致初始库仑效率低、容量快速衰减和电压衰减。所有这些改性策略都是为了突破电化学性能的极限,但很少有研究考虑后处理过程,如循环LMRO的修复或再生。简单的热处理,会导致严重的TM迁移,并导致层状结构缓慢松弛/坍塌到岩盐相,因为循环的LMRO是一种亚稳态结构。相比之下,集中光的集中太阳辐射(CSR)可以在短时间内提供超强辐射和超高温,为有限的副反应修复消耗的LMRO提供了有效的能量。与传统的热退火相比,CSR具有光催化和光热相结合的优点。
成果简介
近日,川大何欣&南科大林苑菁&浙大陆俊团队利用CSR工艺直接、有效地修复了降解的LMRO材料。由于TMs和O-阴离子的氧化还原活性的重新激活,修复后的电极的放电容量成功地恢复到原来的水平。该工作以“Efficient direct repairing of lithium- and
manganese-rich cathodes by concentrated solar radiation”为题发表在Nature Communication上。
研究亮点
LMRO-50C的表面主要由LiMn2O4和MnO2构成,它们可以吸收从紫外到红外线波长的辐射,产生空穴-电子对。因此,它们作为产生空穴的诱导层,实现在晶格中的转移。
此外,电化学循环在LMRO-50C中形成的氧空位可以有效地减轻空穴-电子对的重组,促进空穴转移到粒子内部。
3.CSR还显著抑制了快速容量衰落和电压衰减。反尖晶石相有效地引入粒子中,形成层状尖晶石结构共存。这种独特的结构可以保持相的稳定性,降低内部应变,从而保证了稳定的循环性能。
图文导读
太阳光模拟装置的光路如图1a所示,光通过特殊的菲涅耳透镜矩阵集中,在工作平面上提供均匀分布矩阵图案的集中光点。沿单个聚焦光束垂直方向的光强由光强传感器测量,以提供辐照度细节。在CSR过程中,工作平面位于聚焦点以下5 mm的水平平面(图1a),反应点在短时间内经历了强辐照和高温。在CSR过程中,LMRO电极缓慢移动到水平工作平面,经历直接入射和散射光辐射,以及电极内的热传递,焦点阵中各焦点的中心温度设置为350°C,避免粘合剂和导电炭黑分解。从Li1.014Mn0.56Ni0.15Co0.13O2(LMRO-50C)到Li0.762Mn0.56Ni0.13Co0.11O2(LMRO-50CS),LMRO经历了明显的锂离子损失和Ni,Co略有缺失。通过电感耦合等离子体发射光谱(ICP-OES)定量。SEM图像(图1c、d)显示了CSR处理前后电极的形态学变化。经过50次循环后,LMRO粒子被由电化学分解产物组成的致密的正极-电解质界面(CEI)层所覆盖(图1c)。CSR处理有效地部分去除CEI层,电极的形态没有被破坏。
利用X射线粉末衍射(XRD)测定了LMRO经过循环和CSR处理后的晶体结构演化。原始LMRO(LMRO)、50次循环后LMRO(LMRO-50C)和修复LMRO(LMRO-50CS)的晶体结构差异在图1e的XRD模式中。(003)、(101)和(104)的峰向较低的角度偏移,所有的峰在循环后都被加宽。衍射峰的位移是由于不可逆的锂离子损失和新相(如尖晶石相和岩盐相)的形成引起的。峰的展宽与局部晶格中TMs无序度的增加和畴尺寸的减小有关。随着蜂窝状LiMn6的破坏,20°~25°的特征峰在循环过程中完全消失。虽然先前的研究已经证明,从循环LMRO材料的高温复锂化中可以恢复原始的超晶格结构,但从LMRO-50CS样品的XRD模式中没有观察到超晶格峰。
然而,(220)特征衍射峰(2Theta=30.5°)的出现表明,CSR过程诱导了大量尖晶石结构的产生,并伴随着部分反尖晶石。由于尖晶石LiMn2O4和Li4Mn5O12中Li原子在8a位和Mn原子在16d位的部分占据,在31°左右的(220)衍射峰通常出现弱或掩蔽。在LMRO-50CS样品中,(220)峰的增强归因于反尖晶石结构中局部原子的重排,这类似于LiNiVO4,其中V原子占据晶体结构中的8a位点,Li/Ni原子同样共享16b位点。在LMRO-50CS的情况下,更高的峰强度对应着更多的TM原子迁移8a位,更多的Li原子占据16d位。根据这些表征结果,图1f所示的示意图显示了在电化学过程和CSR处理后,LMRO的组成变化。
图1g和图h中的LMRO-50C和LMRO-50CS的HRTEM图像,详细地展示了局部结构的演化。经过50次循环后,由于不可逆的氧释放和表面重建,在颗粒表面形成尖晶石和岩盐相(图1g)。在CSR处理过程中,光催化和光热效应的结合导致了表面和大块的大尖晶石畴,从而导致整个粒子的激光尖晶石相共存(图1h)。尽管粒子的大部分仍然主要由层状结构构成,但尖晶石颗粒的存在和混合相的存在证明了在CSR过程中存在的实质性相变。
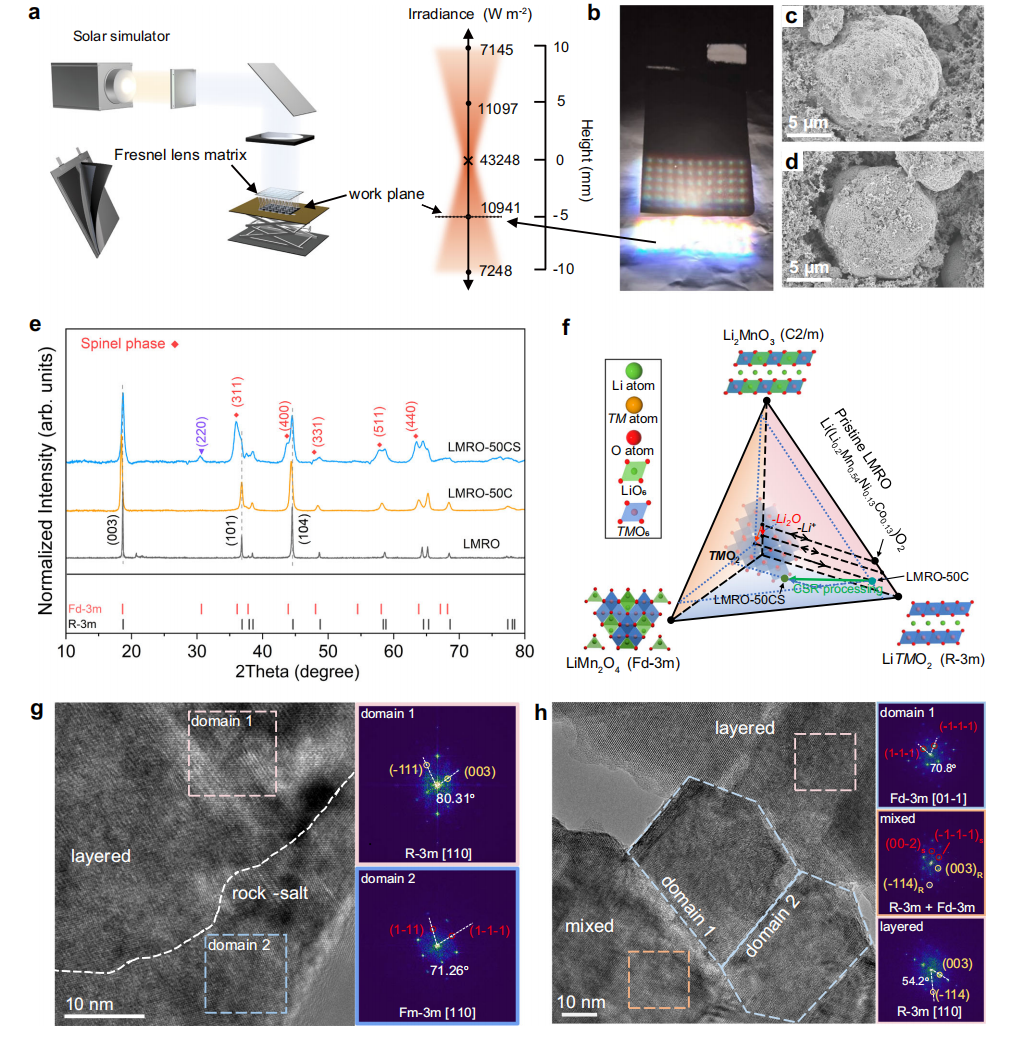
图1. 通过CSR工艺直接修复电极。a太阳光模拟装置的光路及焦点处的光强分布示意图,工作平面的高度(虚线)在焦点以下6 mm。b通过菲涅耳透镜矩阵集中太阳光进行CSR处理下LMRO电极的真实图像。LMRO-50C (c)和LMRO-50CS (d).的扫描电镜图像e LMRO、LMRO-50C和LMRO-50CS的XRD模式,红色标记对应尖晶石相,紫色标记代表CSR处理后产生的反尖晶石相。f电化学循环和CSR处理后相应相的组成变化示意图。LMRO-50C (g)和LMRO-50CS (h)的HRTEM图像,以及相应的快速傅里叶变换(FFT)模式。下标字符S表示尖晶石相,R表示层状相。
在CSR过程中,粒子表面的化学成分和元素价态经历了显著的演化。采用各种表面检测技术来详细探索粒子表面的演化。为了阐明CSR过程中CEI层的反应机理,我们在LMRO、LMRO-50C和LMRO-50CS电极上收集了x射线光电子光谱(XPS)光谱,如图2a-d所示。三个样品在284.8 eV处都有一个较强的C-C峰,这是由于电极中的导电炭黑(Super-P)引起的结果。LMRO-50C样品显示更强的C-O和C=O峰值在286.6eV和288.8eV,和533.6eV和532.6eV 的O1光谱与LMRO相比,属性有机组件ROCO2-LiR-CFx,和R-COxFy,聚碳酸酯在CEI层形成。在F 1 s光谱中,一个相对较高的在LMRO-50C表面观察到无机成分(686.8 eV)、LixPOyFz(685.6 eV)和氟化锂(684.7 eV),这归因于LiPF6盐的分解。氟化锂(55.8 eV)和Li2CO3(55.2 eV)在CEI中的富集也出现在Li 1 s谱中,这与之前的报告的结果一致。与LMRO-50C相比,在LMRO-50CS样品中,C 1 s和O 1 s光谱中观察到的C-O和C=O峰几乎消失。这种消失归因于在CSR过程中由局部高温引起的CEI中有机组分的分解。值得注意的是,由于这些无机组分的热稳定性较好,因此在CEI层中,代表无机组分的峰并没有发生明显的变化。因此,LMRO-50CS样品的CEI层中富含氟化锂、Li2CO3、LixPFy和LixPOyFz等无机组分,有助于改善修复材料的循环稳定性。
TMs阳离子和O阴离子的氧化态揭示了不同状态下电极表面的重构,如图2e-h所示。软x射线吸收光谱(sXAS)光谱采集自LMRO、LMRO-50C和LMRO-50CS样品。经过50个循环后,Mn L3边和Co L3边的低能肩(峰i)的出现都显著增加,表明TM3d与更高的占有率结合,并且在循环时出现了Mn2+和Co2+。随着循环电极的进一步CSR处理,LMRO-50CS的Mn L3边缘的峰i大大减少,而峰iii显著增强,并向较高的能量区域略有移动,显示出与二氧化锰相似的线形。这表明,在CSR处理过程中,锰被氧化为四价物,并伴随着锂离子的萃取。对于Co L3边,峰i的减少与峰ii的高能位移有关,表明Co的氧化。与Mn和Co的氧化不同,由于Ni的氧化电位位于LMRO-50C之外,导致与原始态和LMRO-50C样品相同的价态,Ni即使在CSR过程后仍保持二价。
一般来说,O K边的前边缘谱的强度和线形以未占据的TM3d-O2p杂化态的密度为主,宽键分别对应于TM4sp和O2p的杂化。图2f所示的LMRO的OK边谱与二氧化锰非常相似,具有较强的前边峰,这是由于锰浓度高和Mn4+中有大量未被占用的三维轨道。经过50个循环后,峰值强度的变化可以从两个方面来解释:一是循环过程中Mn/Ni的还原和表面尖晶石/石盐相的形成。其次,电解质过程中分解形成含氧副产物(如氧化锂、Li2CO3、氢氧化锂、RO-Li和ROCO2-Li)。在这些含氧的副产物中,没有可用的轨道与O2p轨道杂交,从而产生较弱的边缘前峰特征。
此外,在536 eV左右的强峰代表了粒子表面CEI成分的存在。值得注意的是,CSR处理后的前边缘模式与TM-O的杂交和表面副产物的分解密切相关。同时,电子从Mn和Co的三维轨道上去除,并导致其氧化。锰和钴的氧化为电子的转移提供了驱动力,O2p到TM波段来补偿电荷的不平衡,这导致了前边缘峰的增加。536 eV时峰值强度的下降是由于CSR处理过程中副产物的分解,这与之前的SEM图像(图1d)和XPS光谱(图2a-d)的结果一致。虽然CSR过程的工作温度已经接近了PVDF的分解温度,但由于快速处理,PVDF粘结剂的分解受到了限制。XPS的结果也证实了这一点,其中LMRO-50CS样品在F 1 s光谱中,在688 eV处仍有一个很强的PVDF代表性峰(图2c)。
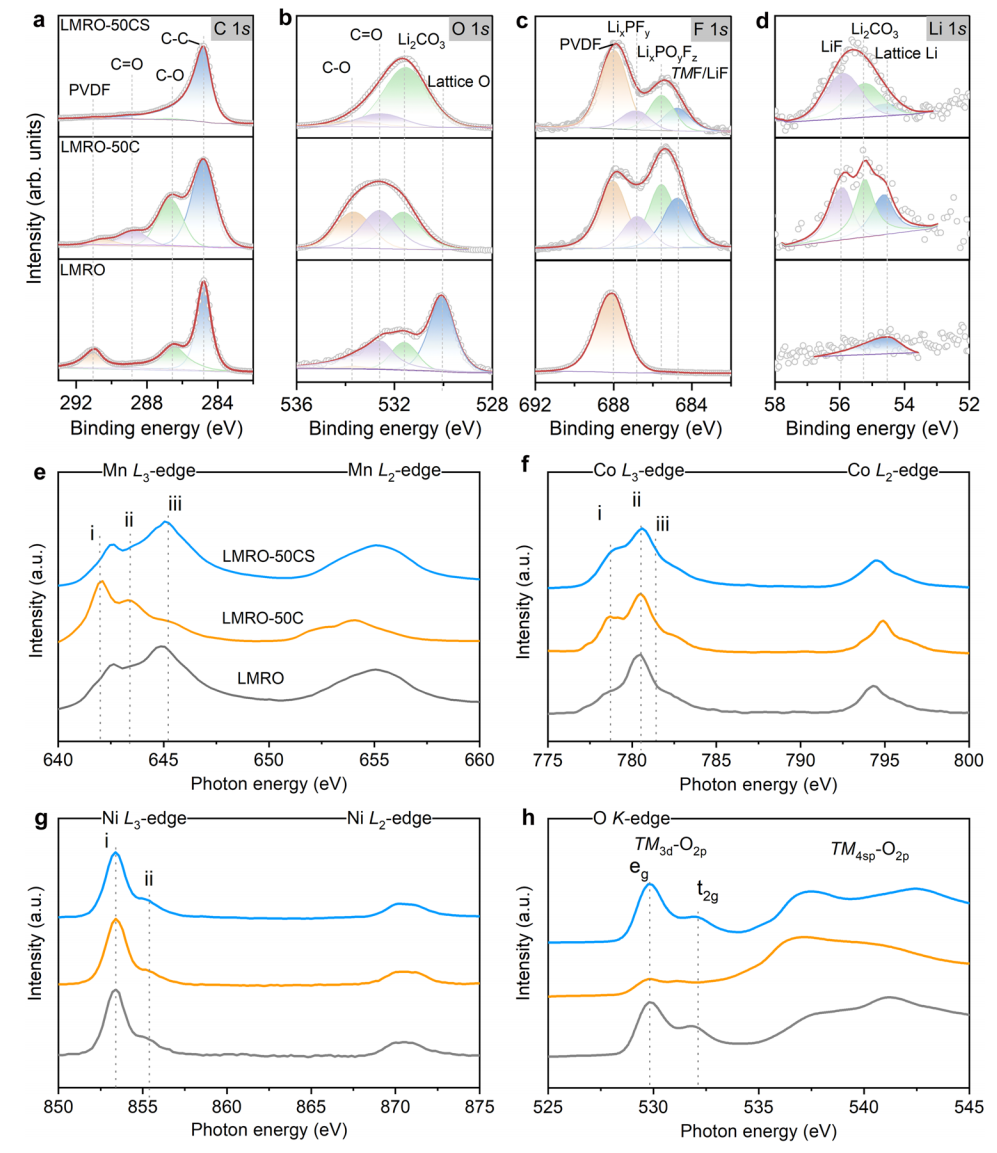
图2. 表面成分从LMRO到LMRO-50C到LMRO-50CS的演变。LMRO、LMRO-50C和LMRO-50CS样品的C 1s (a)、O 1s (b)、F 1s (c)和Li 1s (d)的XPS光谱。在总电子产率(TEY)模式下收集的Mn L边(e)、Co L边(f)、Ni L边(g)和O K边(h)的sXAS模式。
LMRO及其回收电极的电化学性能如图3所示。在C/2(1C=250mAhg−1)下进行50次循环后,常规LMRO的容量降低到220mAhg−1,并在循环过程中继续下降。当在循环电极上直接使用CSR处理时,所输送的放电容量恢复到273mAhg−1,有效地缓解了衰落趋势。然而,从LMRO-50CS样品中也可以观察到一个不寻常的初始循环循环效率,这与修复后的Li0.762Mn0.56Ni0.13Co0.11O2中较低的Li含量相对应。由于晶体结构中存在大量的锂空位,锂离子可以在随后的放电过程中重新插入。在第二个循环中,充电容量达到273mAhg−1,分别在3.0-3.3V和2.9-2.6V的电压范围内出现了一个长长的氧化还原平台。这种低电位平台对应于3.0V的氧化峰和2.8V的还原峰,这通常与在LMRO材料中引入尖晶石相有关。LMRO-50CS在循环过程中的压降可以忽略不计,并且在100次循环后具有高容量保留(图3c)。由于容量快速的衰减和电压的衰减得到了缓解,在长时间循环中,LMRO-50CS可以保持比LMRO更高的能量密度。LMRO和LMRO-50CS的倍率性能比较如图3d。LMRO-50CS在1C之前提供了更高的放电能力,但在高倍率下略低。
第一次充放电过程中获得的非原位XRD(sXRD)谱如图3e所示,(003)峰的位移与晶格c的收缩和膨胀有关,是由脱锂和锂化过程引起的。由于尖晶石相干结构的存在,LMRO-50CS样品显著抑制了(003)位移,有效地抑制了结构畸变,保持了晶格的稳定性,从而提高了循环过程中的容量稳定性和电压稳定性。此外,(220)峰的弱但高度可逆的变化代表了反尖晶石相的强活性。因此,电化学过程是在多相氧化还原反应的混合物下进行的。
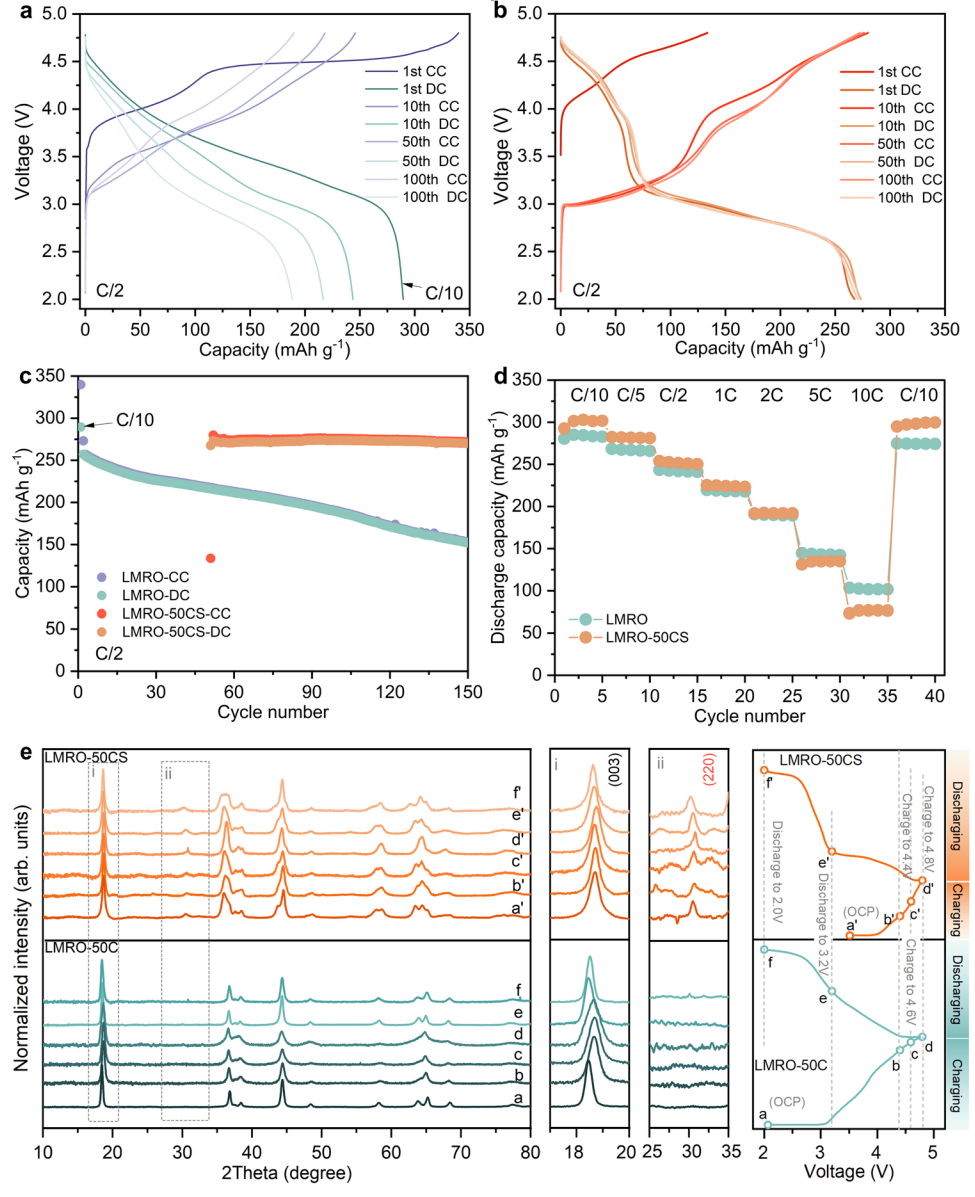
图3. 电化学性能和非原位sXRD测量。LMRO-50C (a)和LMRO-50CS (b)电极的充放电曲线。LMRO和LMRO-50CS的C/2 (c)下的循环性能和倍率性能(d)。e LMRO-50C和LMRO-50CS样品在代表性电压下的非原位sXRD,放大图范围在2Theta=17.5-20°和25-35°范围内。a-c中的(CC)表示充电状态,(DC)表示放电状态。
为了进一步研究电荷补偿的定量表征,采用sXAS在总电电子产量(TEY)和总荧光电电子产量(TFY)模式下测量了电极中TMs的L边(如图4a-c所示)。显著的模式变化表明氧化还原偶从LMRO-50C到LMRO-50CS的严重进化。结合光谱变化和拟合结果,在第50个循环中,定量的氧化还原偶数分别为Mn3.33+/Mn2.77+、Co3.64+/Co2.88+和Ni2.69+/Ni2.14+,如图4d所示。在电化学循环过程中,Ni的活性降低,而Mn的贡献显著增强。Ni氧化还原的较低容量贡献可归因于Ni离子从体向表面迁移和在电解质中溶解。此外,Ni的还原导致更多的Mn和Co的活化,导致循环过程中电压逐渐下降。
由于经过CSR处理后,Mn被氧化至4+,当电压达到4.8 V时,LMRO-50CS样品中Mn没有进一步的氧化。值得注意的是,Co和Ni都没有被氧化为四价物,这与层状LMRO在初始循环中的氧化能力不同。在放电过程中,Ni降低到2+,但Co和Mn降低到比原始水平低得多的水平。当放电到2.0V时,Mn L3边缘的峰ii和峰iii消失,光谱的形状与Mn2+的参考光谱高度一致,这表明几乎所有的Mn4+都被还原为Mn2+。在放电过程中,Co的特征峰随着光谱形状的变化而向较低的能量范围移动,特别是在3.2V~2.0V的低压范围内,在表面有更多的Co3+被还原为Co2+。由此可见,Ni3+/Ni4+、Ni2+/Ni3+和Co3+/Co4+氧化还原主要负责高压斜率(4.8V-4.0V)的反应,而Co2+/Co3+和Mn2+/Mn4+氧化还原参与LMRO-50CS从3.2V~2.5V的放电。
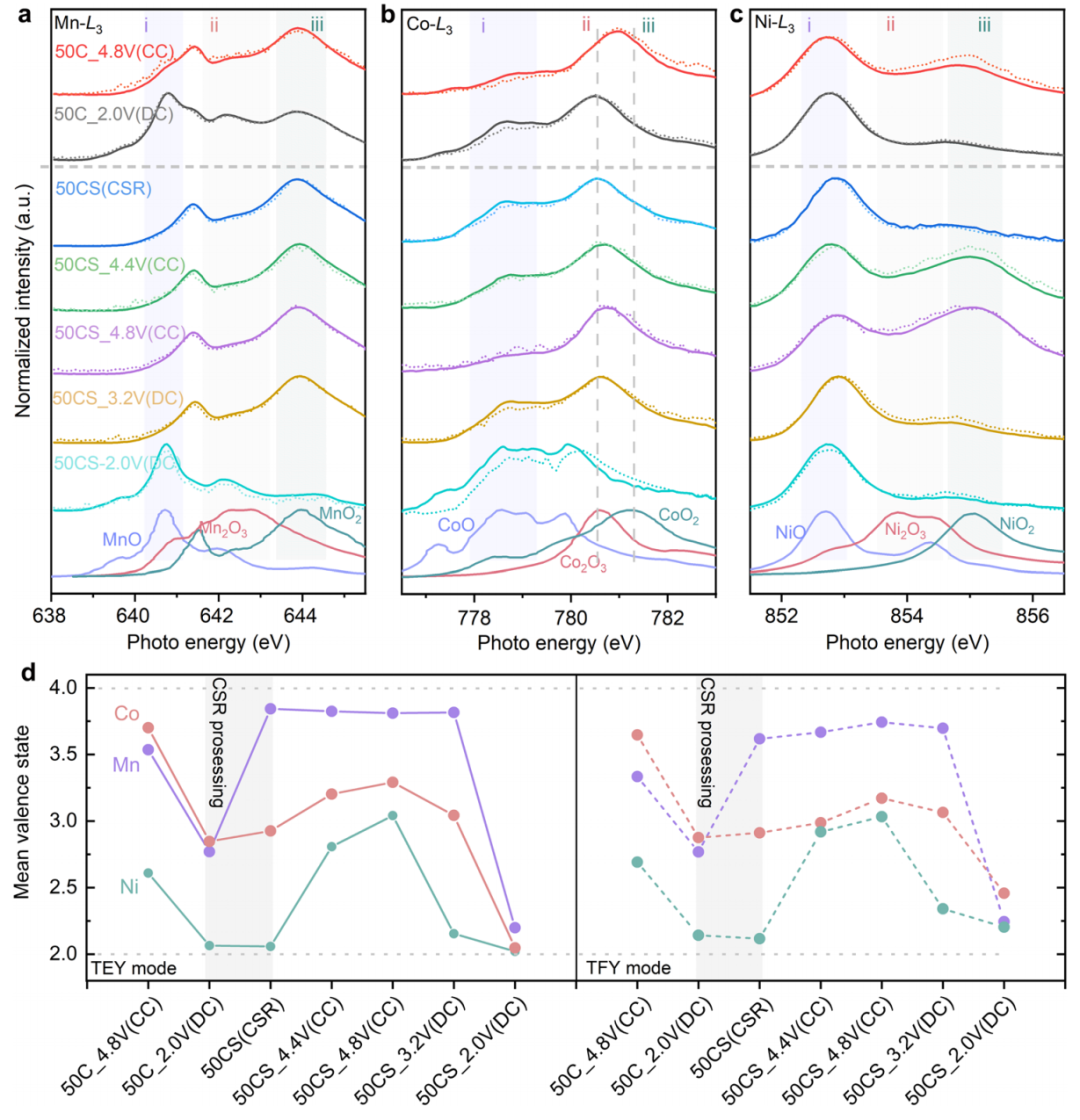
图4. TMs的氧化状态定量分析。a-c在代表电极的TMs(Mn、Co、Ni)-L3边缘TEY(实线)和TFY(虚线)光谱。XAS光谱按最高峰与背景线之间的高度进行归一化处理。d根据光谱中价分布的拟合结果,确定每个TM的量化平均氧化态。(CC)表示充电状态,(DC)表示放电状态,(CSR)表示CSR处理后的样品。
除了TMs的关键作用外,O-阴离子在LMRO-50CS中的贡献在电荷补偿中也具有很高的相关性。CSR处理前后粒子横截面的高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像及相应的EELS图像如图5a和b所示。通过LMRO-50C材料的粒子,可以观察到明显的峰前特征转变,前峰的强度从体积到表面逐渐减弱,最终在梯度深度为20 nm的表面处消失。然而,LMRO-50CS粒子的弱O-峰前区域进一步扩大,并在体积中出现了一个间隔。O前峰的减弱与O空位的形成密切相关。由此可见,可以得出结论,CSR过程形成了大量的O空位。氧空位的引入会降低O2P带的DOS,并降低TM-O键的共价性,从而增强了电荷补偿过程中晶格氧氧化还原的可逆性。
为了量化氧的性能,利用共振非弹性x射线散射(mRIXS)技术在相同的激发能范围内区分固有的氧氧化还原特征和强TM-O杂化特征。在530.5-531.5 eV的激发能范围内,所选区域的mRIXS图如图5c所示。当电极充电到4.8V时,在525eV发射能量左右的垂直和宽杂化特征进一步变宽,在531eV激发和523.7eV发射能量左右出现一个特征,代表氧化氧的出现。从LMRO-50C充电4.8 V、LMRO-50CS充电4.8 V和放电3.2 V的样品中可以识别出氧化晶格氧的特征。为了详细分析,综合mRIXS强度如图5d所示。在523-525 eV发射范围内的峰值可以直接和定量地表示晶格氧氧化。从图5c的mRIXS数据和图5d的综合数据可以看出,LMRO-50C样品充电至4.8 V,放电至2.0 V时出现晶格氧氧化的特征信号。结果表明,晶格氧在50次循环后,虽然其容量贡献持续下降,但仍参与电荷补偿。在LMRO- 50CS充电过程中,充电至4.4V后开始出现氧氧化特征,在4.8V时出现最强峰值。当放电到3.2 V时,这一特征仍然存在,直到放电到2.8 V时才会湮灭。黄色阴影区域(图5d)的定量分析如图5e所示。经过CSR处理后,峰面积的变化范围有所扩大。这一定量结果为CSR处理提高高可逆性氧氧化还原活性的假说提供了强有力的证据。LMRO-50CS的差分电化学质谱(DEMS)与mRIXS的结果一致,在C/5处的第一次电荷过程中释放的氧量可以忽略不计。这为晶格氧氧化还原反应的高可逆性提供了强有力的证据。
基于这些结果,我们可以得出结论,容量的恢复主要来自于TMs和O-阴离子的氧化还原的再活化。由于价态的变化与充放电过程中的容量贡献密切相关,TMs价态变化的增加是容量恢复的主要原因,特别是Mn占LMRO中TMs的大多数。。
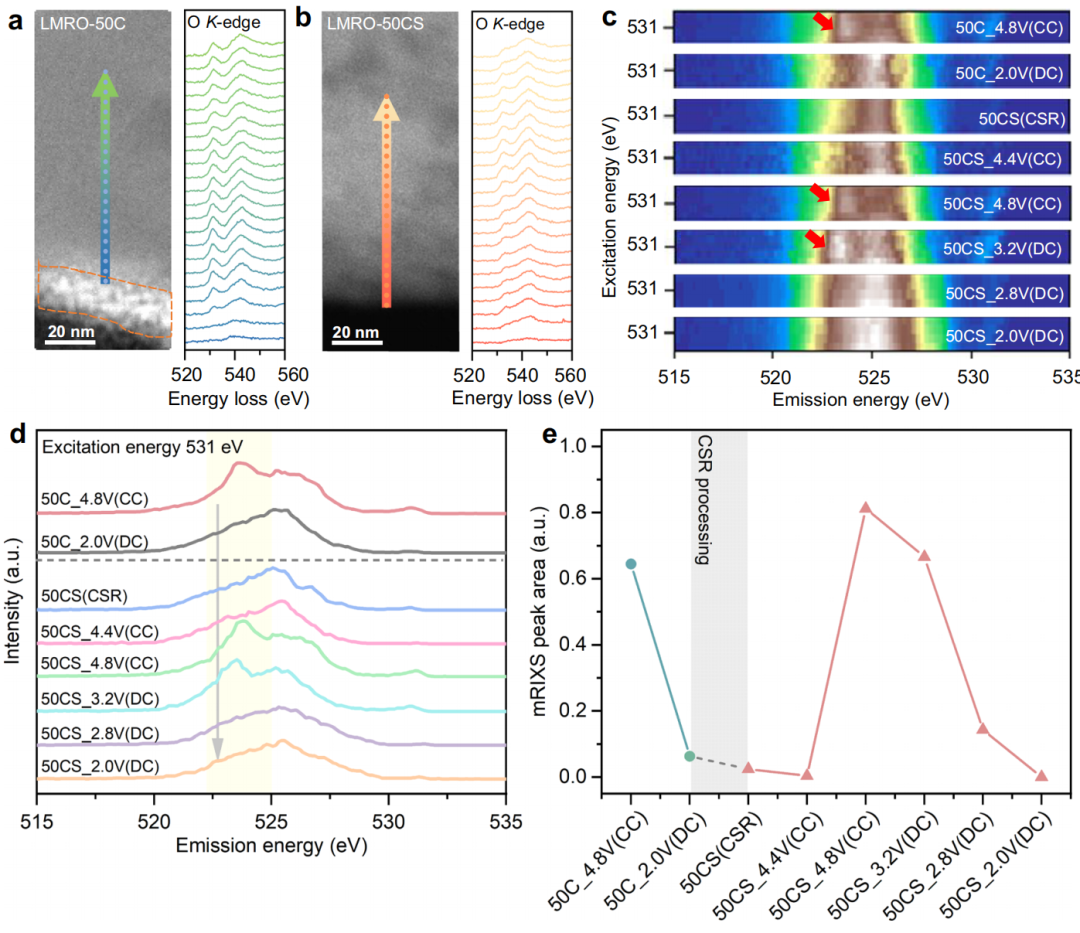
图5. O-阴离子的统计和详细表征。LMRO-50C(a)(虚线内面积为CEI)和LMRO-50CS (b)沿箭头方向的HAADF-STEM和EELS O K边线扫描光谱。c在不同充放电电压下,LMRO-50CS和LMRO-50CS在531 eV左右绘制的局部mRIXS图,红色箭头表示在531 eV激发能和523.7 eV发射能左右出现的氧氧化特征。d特征能量范围内的综合RIXS强度,从530.5到531.5 eV。e对(d).中黄色阴影区域的积分面积的量化(CC)表示充电状态,(DC)表示放电状态,(CSR)表示CSR处理后的样品。
经过电化学循环,在粒子表面产生了LiMn2O4和二氧化锰等尖晶石结构。据报道,LiMn2O4可以对可见光作出反应。在光照下,该材料可以被激发产生微秒寿命的光产生的空穴-电子对,以补偿充电过程。此外,还进一步证明了该材料可以响应从紫外到红外线的较长波长,并且在红光下的响应更强。此外,二氧化锰因其环境友好、成本低、带隙窄、可被可见光和红外光激发,是一种广泛应用的光催化剂。因此,LMRO- 50C复杂的表面结构组成与CSR过程密切相关。为了更深入地了解CSR过程中的反应机理,我们对该材料的光学性质进行了一系列的研究。Tuac图从紫外线和可见分光光度漫反射光谱(UV-Vis-NIR DRS)图6a所示,这表明带隙能量(如)LMRO尖晶石LiMn2O4(LMO)和LMRO-50C分别为1.112 eV,0.836 eV和1.163 eV。通过电化学莫特-肖特基分析计算得到的平带势(Ef)分别为−1.065V(LMRO)、−0.924V(LMO)、−0.792V(LMRO-50C)。因此,LMRO-50C的导通带电位(ECB)可以估计为2.352 V (vs。Li+ /Li).根据EVB = ECB+Eg公式,我们可以进一步得到LMRO-50C的价价带势(EVB)为3.515V。以上述结果为基础,电极的相对势能图见图6c,其中Co2+/Co3+和Mn2+/Mn4+氧化还原电位在LMRO-50C的导带顶部以上,因此可以被氧化,而Ni2+/Ni4+和Co3+/Co4+,O2−/On−在价带顶部以下,不能被光生空穴氧化。
基于上述结果,我们提出了一种光催化氧化-还原过程的机理。由电化学循环引起的表面尖晶石相(LiMn2O4和二氧化锰)组分吸收光能产生空穴和电子,空穴在晶格中转移,并被相邻的低价过渡金属Mn2+、Mn3+和Co2+捕获,它们分别被氧化为Mn4+和Co3+。当TMs被氧化时,锂离子从结构中喷射出来,留下锂空位。在CSR过程产生的热效应下,过渡金属更容易迁移到Li层中的Li空位,形成不可逆的结构相变。强(220)衍射峰证明了形成的尖晶石具有部分无序的反尖晶石结构,其中活性锂离子占据了锰的八面体位置。
除了光催化效应外,光热效应对材料性能的重要影响也不应被忽视。由于CSR处理可以迅速将材料表面加热到约350°C时,热量转移到本体中,加剧电化学循环产生的裂纹,导致材料沿裂纹的有限分解。在热效应下,缺陷层结构分解为尖晶石、岩盐、锂氧化物和氧。各种锂氧化物和氧被转移到粒子表面,释放和蒸发。同时,在300°C以上的高温下,锂离子的损失,导致粒子中残留的过氧基分解,产生氧化物,释放氧气,进一步导致尖晶石结构和O空位的形成。
此外,循环过程中产生的氧空位不仅扩大了光响应范围,还捕获电子或空穴以促进它们的组合。因此,我们推断LMRO材料的光修复性与电极的特殊降解机制有关,它产生了氧空位和一个新的相。我们对初始材料的光处理结果证实了这一点,它没有显示出化学价态和电化学性质的显著变化。考虑到光催化效应的特殊性,粒子需要用足够强度的光照射,并在适当的波长下发生这些CSR反应。此外,在长周期后,LMRO样品中CSR过程的有效修复性降低。
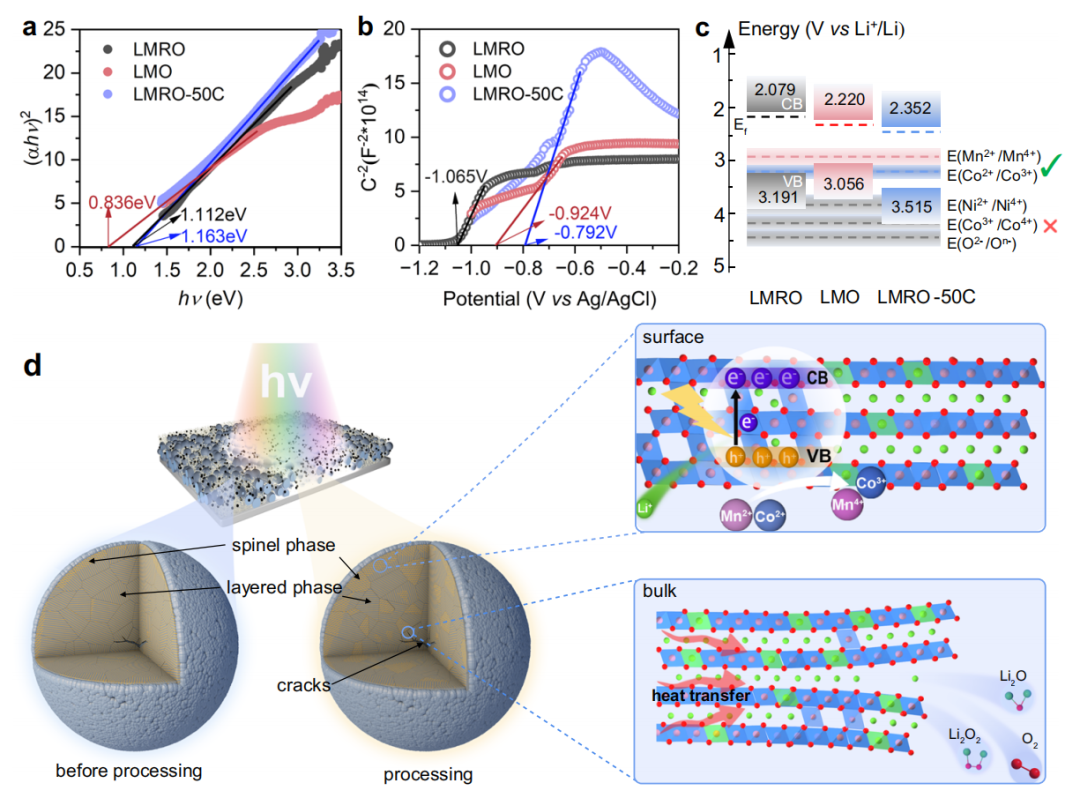
图6. 光催化和光热耦合效应的原理示意图。(αhν)2vs.hν图(a)以及LMRO、LMRO-50C、LMO电极的莫特-肖特基曲线(b)。c由(a)和(b).=计算出的不同电极的导带电位和价带电位的分布。D CSR过程的反应机理图。
总结与展望
综上所述,在本工作中,循环后的LMRO材料可以通过简单、快速、绿色的方法(CSR)处理进行有效修复,修复后的材料具有良好的容量保持率和电压稳定性,探讨了容量恢复的机制,揭示了CSR过程前后电荷补偿机制的演变。sXAS和mRIXS结果表明,在CSR处理过程中,大量的锰被氧化为四价锰,在随后的充放电过程中,TMs(Mn、Co和Ni)和晶格氧的电荷转移数都增加,导致容量恢复。电化学稳定性的改善与两个因素有关:一是氧化还原偶的Co2+/Co3+和Mn2+/Mn4+和晶格氧氧化还原可逆性的增强,而二是CSR处理后红部分反尖晶石相干结构的形成。作者进一步证明了CSR对光催化和光热效应的有益反应与形成的尖晶石相LiMn2O4和MnO2以及充放电循环中的氧空位高度相关。此外,对Co2+/Co3+和Mn2+/Mn4+氧化还原偶的了解将为未来LMRO材料的改性和高能密度电池材料的设计提供可能。
审核编辑:刘清
-
锂离子电池的最新正极材料:掺锰铌酸锂?2016-01-19 6466
-
太阳辐射测量——手持式太阳辐射仪的原理2021-09-07 2504
-
我国研发出富锂锰基动力电池正极材料 1380Wh/kg以上的比能量密度2018-11-23 3282
-
太阳辐射传感器的作用、原理及应用2020-08-24 4270
-
太阳辐射传感器的作用、原理以及应用2020-10-29 2206
-
富镍三元正极材料的改性策略2020-11-02 11497
-
全面剖析富锂锰基正极材料!2021-05-10 45468
-
浅析太阳辐射表的波长范围及应用2021-09-14 832
-
新型梯度“单晶”富锂正极材料2023-02-01 4485
-
锂锰电池的正极材料是由什么组成的?锂锰电池正极材料的优点2023-11-10 2008
-
富锂锰正极电压衰减机理终于讲明白了!2024-03-22 4169
-
高稳定性富锂锰基正极材料2024-12-10 2406
-
如何测试整车太阳辐射?怎么改进车辆的设计以应对太阳辐射?2026-04-24 180
全部0条评论

快来发表一下你的评论吧 !
