

可充电电池中的高熵电解质设计
描述
研究背景
可充电电池领域在高效和可持续能源技术需求的增加的推动下,已经取得了显著的进步。作为可充电电池系统中的关键组成部分,电解质在决定电池可逆性和稳定性方面起着至关重要的作用。然而,当前电解质的离子电导率不令人满意,并且低温性能有限,这大大阻碍了电池的应用场景。高熵电解质(HEEs)由于其解决上述问题的潜力而受到广泛关注。然而,HEEs的概念模糊、缺乏电解质成分筛选和优化的指导,以及HEE对电极-电解质界面影响的明确程度,严重阻碍了HEEs的实际可行性。
成果简介
清华大学深圳国际研究生院康飞宇教授和周栋教授,首次对新兴 HEE 进行了从设计原理到性能优化的调查。我们总结了各类 HEE(包括液体、准固体和全固体 HEE)的离子传输机制和基本性质,并回顾了可充电碱金属(例如 Li、Na)基电池和多价 HEE 的最新进展。HEE 可以本质上增强离子(例如镁、锌)电池系统的性能。特别强调了高熵溶剂化/晶体结构与电池性能之间的相互作用。最后,指出了开发与 HEE 结合的电池的主要挑战,并为未来的突破提供了前景。该工作以“More is better: high-entropy electrolyte design in rechargeable batteries”为题发表在Energy & Environmental Science上。
图文导读
1.引言
熵的概念最初由鲁道夫·克劳修斯在19世纪中叶提出,用来描述系统中的无序性或随机性程度(图1a)。作为热力学第二定律的核心概念,熵在能量存储和转换过程中具有重要意义。高熵材料(HEM)具有吸引人的优点,例如易于形成具有简单晶体结构的单相固溶体和定制的功能特性,在热电、热能和环境保护、电化学储能等领域获得了极大的关注。催化系统等HEM的演变如图 1b 所示。2004 年高熵合金的出现引发了对其机械性能的广泛研究,其结构通过掺入高度无序元素的最大化构型熵而稳定。2010 年,Kao 等人在高熵合金中观察到晶格畸变,通过创造更有利的反应位点,使 HEM 成为储氢的有希望的候选者。HEM 中功能单元之间的强大协同效应有助于高效的能量转换过程,从而引发了对高熵贵金属和非贵金属的研究用于甲醇氧化、析氧和还原等应用的电催化剂。
随后,高熵氧化物的发现促进了 HEM 在电化学储能领域的蓬勃发展。例如,Wang 等人描述了一种含锂熵稳定的氟氧化物,即Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OFx),由于其3.4V的高工作电位,可用作锂离子电池(LIB)的正极活性材料以及锂存储性能的改进。熵稳定还允许通过以前所未有的方式改变组成元素来定制电池循环性能。此外,熵稳定的概念可以扩展到具有岩盐结构的氯氧化物。Patra等人报道了一系列尖晶石型高熵氧化物作为LIB负极,通过将附加元素(即Mg、V和Cu)掺入四元中熵成分(CrNiMnFe)3O4中,其中Mg充当结构支柱, Cu 降低了成本,V 有利于提高电池容量。所开发的 (CrNiMnFeCu)3O4 显示出更高的倍率容量。
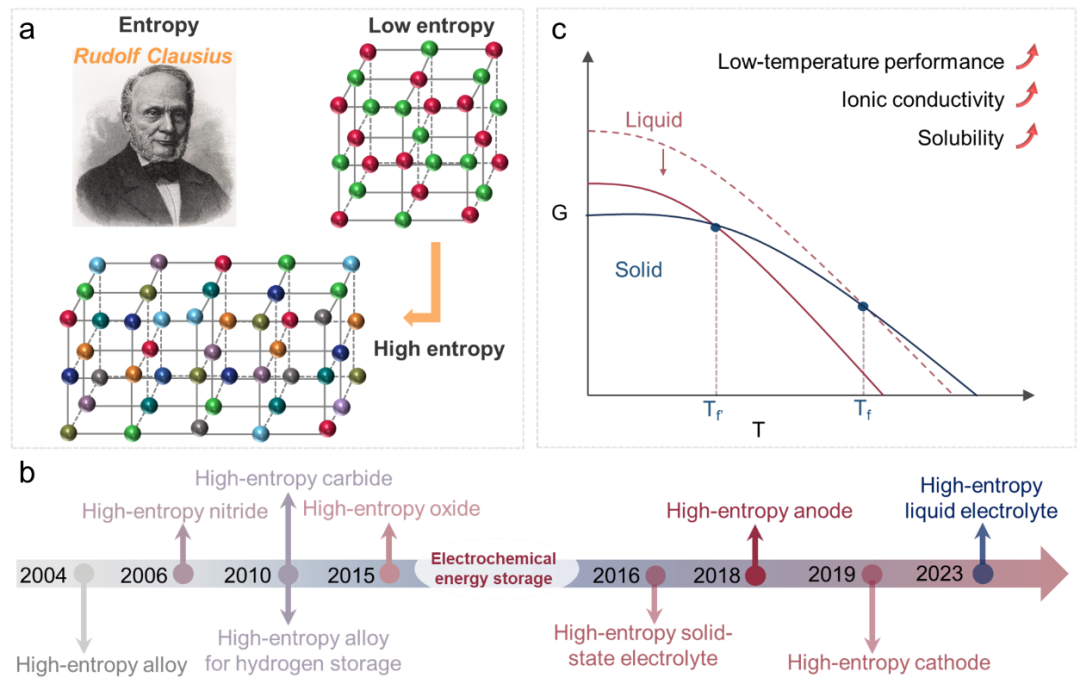
图1 HEE的主要进展和优势总结。(a) 熵的提出。(b) HEM 和 HEE 的发展时间表。(c) HEE 中冷冻温度 (Tf) 的高熵效应和其他高熵影响的机制。
尽管HEM在能源相关应用中引起了相当大的兴趣并经历了快速增长,但用于可充电电池的高熵电解质(HEE)的进展仍处于起步阶段。HEE 系统日益复杂,使得电池性能的预测和定制变得更具挑战性,因为即使电解质成分或化学计量的微小变化也可能显着影响电化学反应的可逆性。尽管困难重重,最近一些开创性的工作揭示了HEE在可充电电池中的巨大应用潜力。一般来说,用于可充电电池的HEE可分为四大类,包括非水液体HEE、水性液体HEE、全固体和准固体(即凝胶)聚合物HEE以及全固体无机HEE。Berardan等人报道了一种高熵固态电解质(SSE),即(MgCoNiCuZn)1−xAxO(A=Li,Na),其中Li或Na可以取代岩盐结构,从而形成氧空位。当x从0.02增加到0.33时,(MgCoNiCuZn)1−xAxO 的Li+电导率与Li+离子的数量成正比。
同时,非水和水HEE已开发用于可充电电池。然而,基于对高熵效应的深入理解来合理设计HEE仍然是一项艰巨的任务。在这里,我们首次对 HEE的最新进展进行全面回顾,从非水和水性液体 HEE、全固体和凝胶聚合物 HEE,一直到全固体无机 HEE。我们回顾了高熵效应对电解质关键性能的影响,例如离子电导率、热稳定性、电化学窗口和电极兼容性。我们强调了HEE在可充电碱金属(例如锂、钠)基电池和多价离子(例如镁、锌)电池系统中的优点,并介绍了 HEE 在这些电池系统中的剩余挑战和前景。
2. 高熵电解质的基本概念及优点
最佳电解质需要优异的化学和电化学稳定性以及熟练的离子传输能力。吉布斯自由能可以普遍量化所有电解质系统的热力学特征,无论其成分如何,这是通过从系统的焓(ΔH)中减去温度(T)和系统熵(ΔS)的乘积来确定的:
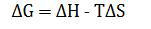
根据上述定义,吉布斯自由能由 ΔH 和 ΔS 之间的竞争决定。通过引入多种成分来增强ΔS,可以减轻ΔH增加的影响,从而降低吉布斯自由能,从而提高电解质的稳定性和反应速率(图1c)。因此,HEE是通过集成不同的组件来实现的,以实现更高的无序性和复杂性。HEE 的灵活性为优化离子传输电导率、电极/电解质界面特性和电池稳定性提供了巨大潜力。值得注意的是,新兴高效率经济体中的“熵”概念仍然含糊不清。最常见的是,它指的是构型熵(Sconf),它是描述混合系统中的不确定性或无序性的物理量。随着 Sconf 的增加,离子可以更自由地移动,更快地适应电场的变化,从而离子的传输速率会增加。同时,Sconf 也会影响电解质的电容性能,因为更高的 Sconf 会导致电解质中的电荷分布更均匀。然而,当 Sconf 太高时,离子的取向和配位可能会变得不稳定,从而降低稳定性,抗极化能力和电解质的耐久性。因此,合理调节 Sconf 对于实现最佳电解质性能非常重要。
超熵 (Sex) 是了解液体 HEE 性质的另一个重要指标,包括溶剂-溶质相互作用、聚集行为和离子动力学等。超熵是实际混合物与理想混合物熵之间差异的结果。电解质的溶解度随着液体电解质中的性增加而升高。同时,性的增加使得电解质更容易结晶,从而降低可逆性和寿命。除了 Sconf 和 Sex 之外,其他形式的熵也被应用于HEE研究中,包括混合熵(Smix)、能量熵和反应熵等。混合熵(Smix)是指不同组分时发生的熵变化混合形成混合物,根据理想解模型或具有相似特性的模型进行计算。Smix用于描述电解质的复杂性。Smix的存在会影响电解质中离子的流动和扩散。能量熵是指由于电解质浓度差异而引起的能量分布的紊乱。能量熵的存在不仅导致液体电解质中离子间能量分布不均匀,影响离子传输速率,而且还调节电解质与溶剂之间的相互作用。反应熵描述了反应前后离子无序程度的变化。化学反应。负反应熵会增加反应的活化能并降低反应速率。HEE 已成为可充电电池领域有前途的候选者。与含有少量成分的传统电解质不同,具有多种成分的 HEE 引入了一种新的范例,用于设计具有增强本能特性和电池性能的电解质。一般来说,HEE 解决传统电解质局限性的优点包括:
金属盐的溶解度增强
传统的液体电解质通常由几种溶剂中的一种与少量作为溶质的单一金属盐组成。在这种情况下,溶质和溶剂之间的相互作用使电解质体系具有有序性增加和结晶的趋势。然而,根据方程(2),多个分量的引入增加了局部无序性,导致更高的Sconf:
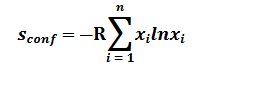
其中R表示理想气体常数,xi表示第i种组分的摩尔分数。改进的 Sconf 降低了溶质结晶的趋势,导致更复杂的溶质-溶剂相互作用和更高程度的离子运动自由度,从而增加了盐在溶剂中的溶解度。
离子电导率改善。
对于液体 HEE,引入具有不同尺寸和电荷的多种离子种类会创建一个复杂的网络,从而驱动较大的 Sex 值:
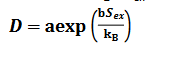
其中D指扩散系数,a和b是通过实验得到的拟合系数,而kB代表玻尔兹曼常数。这种大的性增加了电荷载流子与电解质中其他离子碰撞的机会,提供更多的传输路径并促进扩散过程。这种改进的离子传输效率导致离子电导率增加并减少电池极化。对于由多种元素组成的全固体无机 HEE,结构中缺乏长程有序会减少离子结合,促进离子迁移和导电性,这主要是由于以下三个关键驱动力:
(1)离子扩散路径的多样性。全固态无机HEE由多种离子组成,其晶体结构不存在明显的有序排列。结果,离子在材料内的扩散路径变得更加多样化。这使得离子可以通过不同的通道和位置迁移,减少离子之间的相互阻碍,促进离子迁移速率。
(2)离子结合能降低。在具有长程有序结构的晶体中,离子往往形成稳定的排列,从而产生更强的离子结合能。相比之下,缺乏长程有序的高熵 HEE 中离子之间的离子结合能要低得多,使得离子更容易解离和迁移。
(3)熵效应。由于存在大量不同类型的原子或离子取代位点,HEE系统具有更微观的状态和更高的Sconf。这可以促进离子的无序运动,降低活化能,有利于离子迁移。显示出更高的倍率容量。
优化低温电池性能
共晶效应是指某些混合物中熔点明显低于其组分总和的现象。这是由离子盐和非离子物质(通常是氢键受体)之间独特的相互作用引起的,导致混合物中离子和分子之间形成高度稳定的结构并降低熔点。因此,对于液态HEEs,多种成分有利于减少电解质熔化和拓宽液体范围。同时,HEE中的不同组成和溶剂化结构可以通过调节离子的去溶剂化及其在固体电解质界面(SEI)中的传输来改变电极的界面性质并增强低温下的电极反应动力学。所有这些都促进了液态HEE在低温下的电池性能。对于全固体无机HEE,多重掺杂金属元素能够降低离子排斥力并增加离子/空位的Sconf,不仅增强结构稳定性,而且促进其在低温应用中的电池动力学。上述优点使得HEE对于二次电池的发展极具吸引力。值得注意的是,应用于便携式设备的市售LIB中的液体电解质配方通常由超过5种成分组成,而大多数实验室LIB电解质配方由少于3种成分组成。因此,HEE的研究无疑促进了电解质设计理论的进步,有助于弥合实验室研究和商业应用之间的差距。
3.非水液体高熵电解质
对于非水HEE,与固体电解质相比,液体电解质缺乏分子排列水平上的长程有序性,定量熵测量和比较相对复杂。此外,液体电解质的熵变与其溶剂化结构(例如溶剂化壳中溶剂、阴离子、簇等的类型、比例和分布)有很强的相关性。因此,熵可以作为分析液体电解质特定溶剂化结构的尺度。1986 年,Marcus 构建了熵理论来评估离子熵效应的影响。Karcher 等人将 Marcus 的理论应用于Na离子电解质溶剂化结构的测量。计算并测量了 Na+ 溶剂化壳的溶剂固定熵 (ΔimmobS) 和相应的电解质溶剂冻结熵 ((ΔfS),并且这两个量的比率给出了Na+第一溶剂化壳层中溶剂量的估计。该值在二甘醇二甲醚(DG)中约为1.7,在碳酸亚丙酯(PC)中约为2.3。此外,他们还报道了电解质熵变化极大地影响了电极反应的自由焓,从而导致反应的氧化还原电位发生变化。当应用于可充电电池时,可以通过针对溶剂和盐改变构型熵来实现非水液体HEE,下面分别进行说明。
3.1 锂基电池多溶剂高熵液体电解质
传统的液体电解质通常由几种溶剂和盐组成,结构简单、熵低。相比之下,高熵多溶剂电解质产生更复杂的结构和更高的 Sconf。这种高度混合的状态增加了电解质分子的排列,增强了系统的自由度,有利于提高电极动力学和低温电池性能。同时,液体电解质的溶剂化效应对电化学性能起着关键作用。之前的许多研究都使用焓作为指标来研究Li+与溶剂和阴离子的相互作用。然而,熵也可以用作分析溶剂化结构的旋钮。弱溶剂化液体电解质通常具有优异的抗氧化性能和与负极良好的相容性。
然而,这些都是以牺牲低温下的离子电导率和电极动力学为代价的。高熵液体电解质可以由于溶剂化结构的多种增加而减弱锂离子与溶剂/阴离子之间的相互作用,从而改善电解质低温下的电导率和电极动力学,同时继承了弱溶剂化电解质的电化学稳定性。通过使用优化的溶剂和/或盐设计 HEE 可以实现更高的 Sconf,这有助于平衡电解质系统的焓和熵,从而增强电解质溶解度和减少离子簇。Kim 等人通过增加电解质中溶剂分子的多样性,设计了一系列 HEE溶剂,包括三种线性醚(即二甲氧基甲烷 (DME)、二甘醇二甲醚 (DEGDME) 和二乙氧基乙烷 (DEE)),以及氟化 CO -溶剂(即1,1,2,2-四氟乙基2,2,3,3-四氟丙醚(TTE)和双(2,2,2三氟乙基)醚(BTFE))用于制备三种电解质:1 M DME-TTE (EL2) 中的双(氟磺酰基)亚胺锂 (LiFSI)、DME-DEE-DEGDME-TTE (EL4) 中的 1M LiFSI 和 DME-DEE-DEGDME-TTE-BTFE (EL5) 中的 1M LiFSI。在EL5的弱溶剂化HEE中,高熵效应有效减少了离子团聚,同时保持了特征性的富含阴离子的溶剂化结构(图2a)。
所有三种电解质均表现出富含阴离子的溶剂化结构。第一个溶剂化壳包含大约三个阴离子和一个溶剂分子。尽管微观溶剂化结构没有显着差异,但在纳米尺度上可以观察到介观差异。同步加速器广角 X 射线散射 (WAXS) 非常适合研究介观溶剂化结构,例如簇和网络。随着溶剂数量的增加,观察到离子簇尺寸的减小。这种现象归因于溶剂化熵的升高,使热力学平衡转向有利于Li+和溶剂之间的相互作用,同时抑制离子聚集。EL5在三种电解质中表现出最大的扩散系数,这可以通过同步加速器广角X射线散射(WAXS)的散射矢量从EL2到EL5电解质的逐渐减小来验证(图2b)。考虑到非等温电池的温度系数与熵变之间的正相关关系,EL5 的溶剂化熵达到最大值(图 2c)。这导致 EL5 电解质具有高离子电导率(25 ℃时约为 3.60 mS cm-1),超过了 EL2(约 1.70 mS cm-1)和 EL4(约 1.95 mS cm-1)电解质的值。在2C倍率下,EL2和EL4无负极NMC532-Cu软包电池的初始容量有限,并且会快速衰减,而EL5可以以70%的容量保持率循环近80次循环。由于碳酸亚乙酯(EC)溶剂的高介电常数及其在石墨正极中的无共嵌入行为,因此广泛用于商业锂离子电池。
然而,EC的高凝固点(36.4℃)强烈限制了LIB的低温应用。Zhang等人开发了一系列 EC 含量固定为 10% 但溶剂成分数量不同的液体电解质(由“x mix”电解质中的“x”表示)。电解质中溶剂种类的增加导致溶剂化熵增加。这反过来又导致电解质粘度和凝固点降低。计算和实验结果表明,商业二元(C1:EC-DMC(碳酸二甲酯)中的 C1:1 M LiPF6,C2:1 M LiPF6在EC-DEC(碳酸二乙酯)或三元(C3:1 M LiPF6 在 EC DMC DEC)电解质在-60 oC时完全凝固,而电解质的凝固点随着溶剂组分数量的增加而降低(图2d)。十进制溶剂基 HEE(10 种混合物)表现出 130 oC 的超低凝固点(图 2e)。当与LiMn2O4 |Li4Ti5O12电极配合时,10种混合基锂离子电池在-40 oC下具有80%的高容量保留率,并且在-60 oC下具有超过40 mAh g-1的比容量(图2f)。总而言之,高熵多溶剂电解质创造了具有更高构型熵的更复杂的结构。这种高度混合的状态增加了电解质分子的排列,增强了系统的自由度,有利于提高电极动力学和低温电池性能。
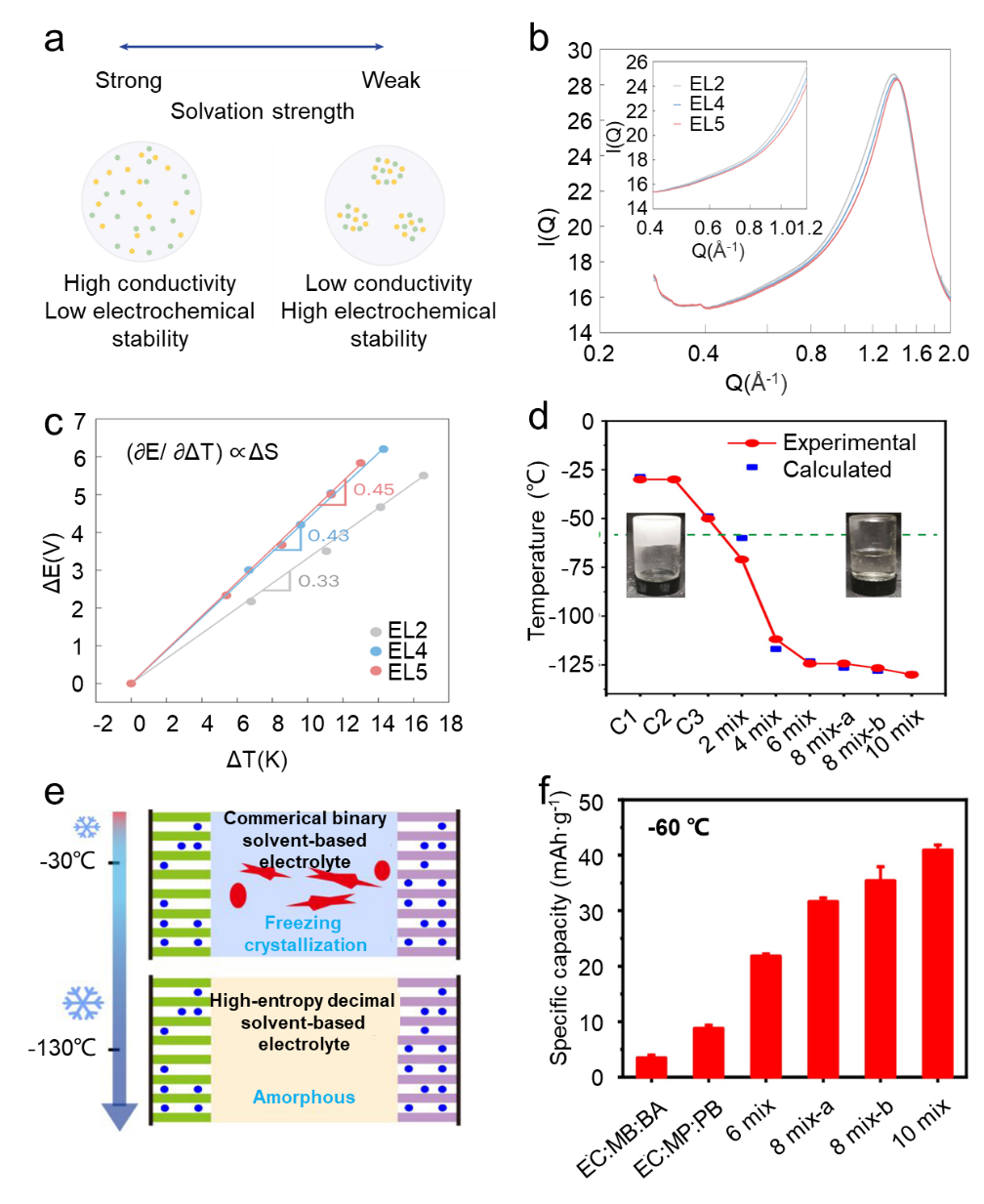
图2 用于锂基电池的多溶剂高熵液体电解质。(a) 调整液体电解质中的溶剂化强度时离子电导率和电化学稳定性之间的权衡示意图。(b) WAXS 显示,当增加电解质中溶剂分子的多样性时,聚类会减少,分子多样性会增加。(c) 电解质的温度系数(由线的斜率表示)。(d) 电解质的实验和计算冰点。(e) 商业电解质和十进制 HEE 的低温行为的示意图。(f) LiMn2O4|Li4Ti5O12 电池在-60 oC 下使用具有不同溶剂量的电解质的容量保持率。MB:丁酸甲酯;BA:乙酸丁酯;MP:丙酸甲酯;PB:丁酸丙酯。
3.2 锂基电池用多盐高熵液体电解质
高熵多盐电解质含有多种盐。这种复杂的盐体系增强了电解质中离子的运动,有利于电解质稳定性的提高。LiNO3作为常用的电解液添加剂,可以有效改善锂金属负极与电解液的相容性。然而,LiNO3 在碳酸酯溶剂中的低溶解度限制了其应用的普适性。这个问题可以通过采用高熵工程策略来解决。为了使 LiNO3 添加剂溶解到碳酸盐基电解质中并形成稳定的均质电解质体系,Wang 等人通过引入多种锂盐来增强电解质的构型熵。有趣的是,每种锂盐对促进 LiNO3 的溶解没有任何作用,而多种锂盐产生的独特溶剂化结构将电解质的熵增加到提高LiNO3的溶解度(图3a)。LiNO3 在所开发的 1.4 M HE-EDF 中的溶解度达到 0.1 M(0.1 M LiFSI/0.1 M 双(三氟甲磺酰基)亚胺锂(LiTFSI)/0.1 M 二氟(草酸)硼酸锂(LiDFOB)/1 M六氟磷酸锂 (LiPF6) 的 EC/碳酸二甲酯 (DMC)(重量比 1:1)与 5% 氟代碳酸亚乙酯 (FEC) 溶液。
此外,通过比较0.75 M HE-PDF(0.15 M LiPF6/0.15 M LiFSI/0.15 M LiTFSI/0.15 M LiDFOB/0.15 M LiNO3 in PC/DEC (1:1) 含 5% FEC )和 0.75 M LiPF6-PDF(0.75 M LiPF6,溶于 PC/DEC,含 5% FEC)电解质的离子电导率,研究了构型熵对电解质温度特性的影响。,适用于宽温度范围。0.75 M HE-PDF的离子电导率显着高于0.75 M LiPF6-PDF(图3b),表明多种盐导致的熵增加有利于加速离子转移。Li|0.75 M HE-PDF|Cu 电池在-40 oC 下循环时保持高于 80% 的库仑效率 (CE),而 0.75 M LiPF6PDF 的 CE 仅为 30%(图 3c)。1977年,Rosenfeld提出了系统内熵与动态特性之间的关系。发现自扩散系数随着超熵的增加而增加。Wang等人。
进一步证明,高熵给电解质带来了高扩散系数。如图 3d 所示,作者开发了 0.6 M HE-DME 电解质,由 0.15 M LiFSI、0.15 M LiTFSI、 0.15 M LiDFOB 和 0.15 M LiNO3 共溶解在 DME 中。更多阴离子基团参与低盐浓度电解质的溶剂化结构;相比之下,该 HEE 中 Li+ 与溶剂和阴离子基团的相互作用要弱得多。这表明通过引入多种盐来提高混合熵提供了调整电解质性能的通用策略。分子动力学(MD)模拟结果表明,0.6 M HE-DME电解液的均方根位移高于0.6 M LiFSI-DME电解液,锂离子扩散系数提高至2.3 × 0.6 M HE-DME 电解质中的 10-6 cm2 s-1 与 0.6 M LiFSI-DME 电解质中的比较(1.2 × 10-6 cm2 s-1,图 3e)。0.6 M HE-DME 电解质的 Li 化学位移表明屏蔽较弱,其溶剂化甚至比稀释的 0.05 M LiFSI-DME 电解质更弱(图 3f)。使用操作数固态 7Li NMR 来量化 0.6 M HE-DME 电解质与 Li 负极的相容性。这使得可以通过奈特位移将锂金属与SEI中的反磁性锂物质区分开来,从而量化剥离后死锂金属的量。结果表明,0.6 M HE-DME 电解质产生较少的死锂和更致密的锂金属形态。总体而言,多盐液体 HEE 增加了阴离子种类的多样性,导致溶剂化结构更加多样化,并削弱了锂离子-溶剂/阴离子相互作用。这可以促进溶剂中某些不溶性盐的解离,并增加离子扩散系数和动力学性质。
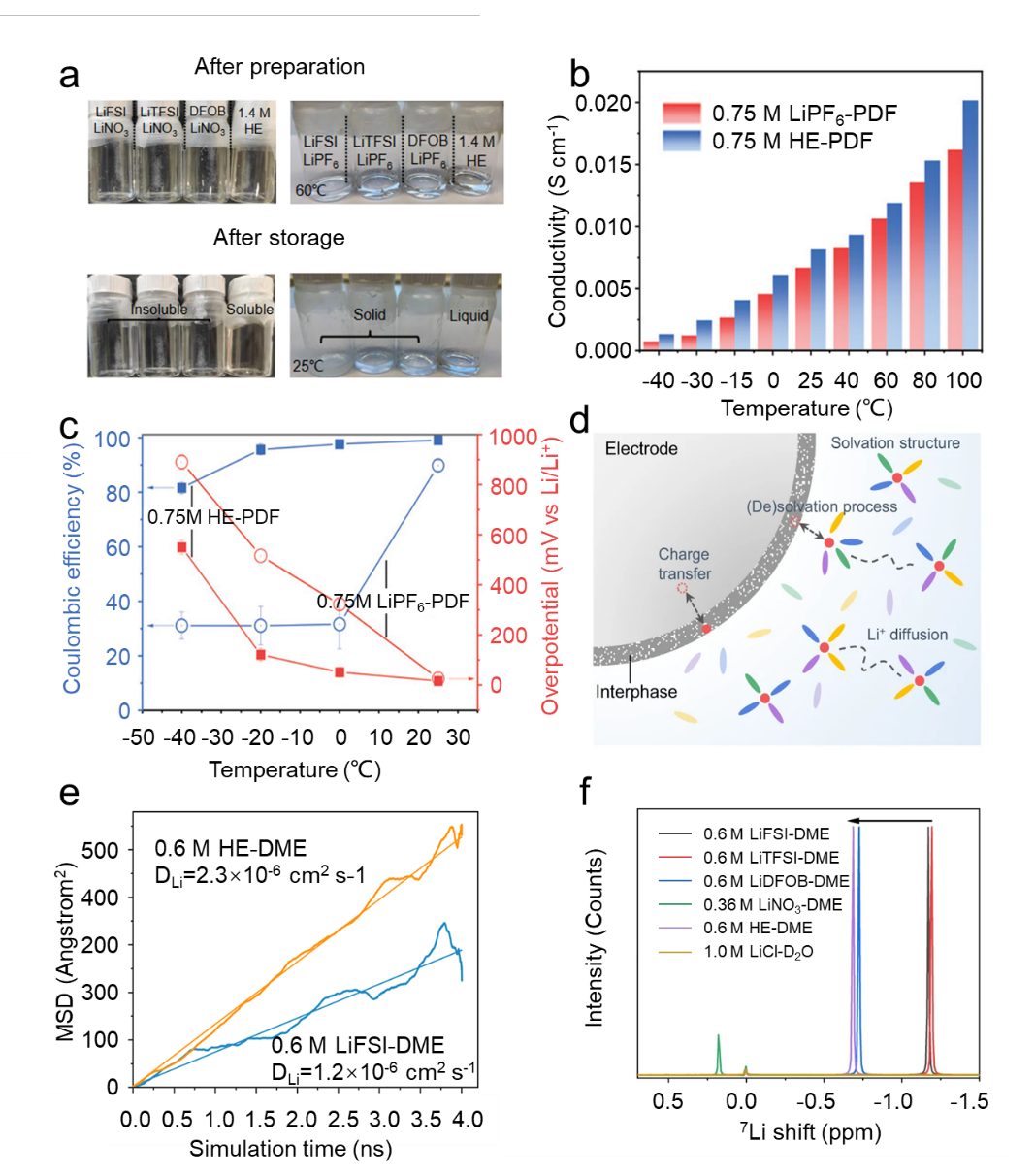
图3 用于锂基电池的多盐高熵液体电解质。(a) 不同盐在 EC 基电解质中的溶解度和固液相变。(b) 不同温度下 HEE 的离子电导率。(c) 在不同温度下使用不同电解质的 Li||Cu 电池的 CE 和过电势。(d) HEE的溶剂化结构和界面行为示意图。(e) 通过 MD 模拟计算的扩散系数。(f)单盐电解质和所制备的 HEE的 7Li 核磁共振 (NMR)谱.
3.3 非锂电池用非水液态高熵电解质
高熵工程策略可以扩展到可充电非锂电池电解质的溶剂化结构设计,包括碱金属(例如钠)基电池和多价离子(例如镁)电池。为了寻找钠离子电池的低温电解质,Yang等人调整了电解质的熵,构建了一种由六氟磷酸钠(NaPF6)在强溶剂化醚(即二甘醇二甲醚(DIG))中组成的混合电解质和弱溶剂化的乙醚(即四氢呋喃(THF))。对于DIG或THF单一溶剂的电解质,随着温度降低,盐析出和沉淀发生,伴随着强离子偶极相互作用,而在二元溶剂电解质中,强离子偶极相互作用和弱离子偶极相互作用在温度变化过程中自适应调节,从而拓宽了离子偶极相互作用。电解质的液相温度范围(图4a)。三种电解质中溶剂分离离子对 (SSIP)、接触离子对 (CIP) 和聚集体 (AGG) 的熵变,即 NDT(DIG 和 THF 中的 1.0 M NaPF6)、NT(THF 中的 1.0 M NaPF6)和 ND(DIG 中的 1.0 M NaPF6)通过 MD 计算。当温度降至-40 oC时,更多的SSIP形成CIP。同时,与弱溶解的NT和强溶解的ND电解质相比,NDT电解质的熵增加,并且没有形成导致熵减少的AGG(图4b)。与ND和NT电解质相反,NDT中即使温度降至-96℃,接近THF溶剂的凝固点(-108.5℃),也不会发生盐沉淀(图4c)。在NDT电解液中,Na+与DIG分子螯合,导致DIG分子的自由度降低,而仅通过一个氧原子与Na+配体结合的THF分子保持自由旋转。
随着温度降低,更多的THF分子参与溶剂化过程,有利于NDT电解质的熵增加并避免盐沉淀。这种高熵溶剂化结构设计为提高可充电钠离子电池的低温性能提供了一种实用的方法。多价离子电池中也有 HEE 的报道。王等人。开发了一种用于镁离子电池的多组分 HEE,含有三氟甲磺酰亚胺镁 (Mg(TFSI)2)、DME、三氟甲磺酸锂 (LiOTf) 和磷酸三甲酯 (TMP),以稳定镁金属负极。纯Mg(TFSI)2/DME 电解液中静电势的正电荷富集均匀分布在DME 分子的C-O 键上。LiOTf和TMP的添加导致溶剂化结构发生显着变化,这一点经NMR证实(图4d)。其中一个溶剂化的DME分子被OTf-取代,桥接Li+形成Mg2+-2DME-OTf--Li+-DME-TMP溶剂化结构(图4e)。这种高熵溶剂化结构重新分配静电势以减轻DME的还原,并优化去溶剂化过程并在负极表面形成富含Mg2+的Mg3(PO4)2导电层(图4f),从而提高了沉积/溶解镁的可逆性。HEE设计为多价离子电池电解质设计开辟了一条新途径,有望解决严重的离子共轭和界面钝化问题。
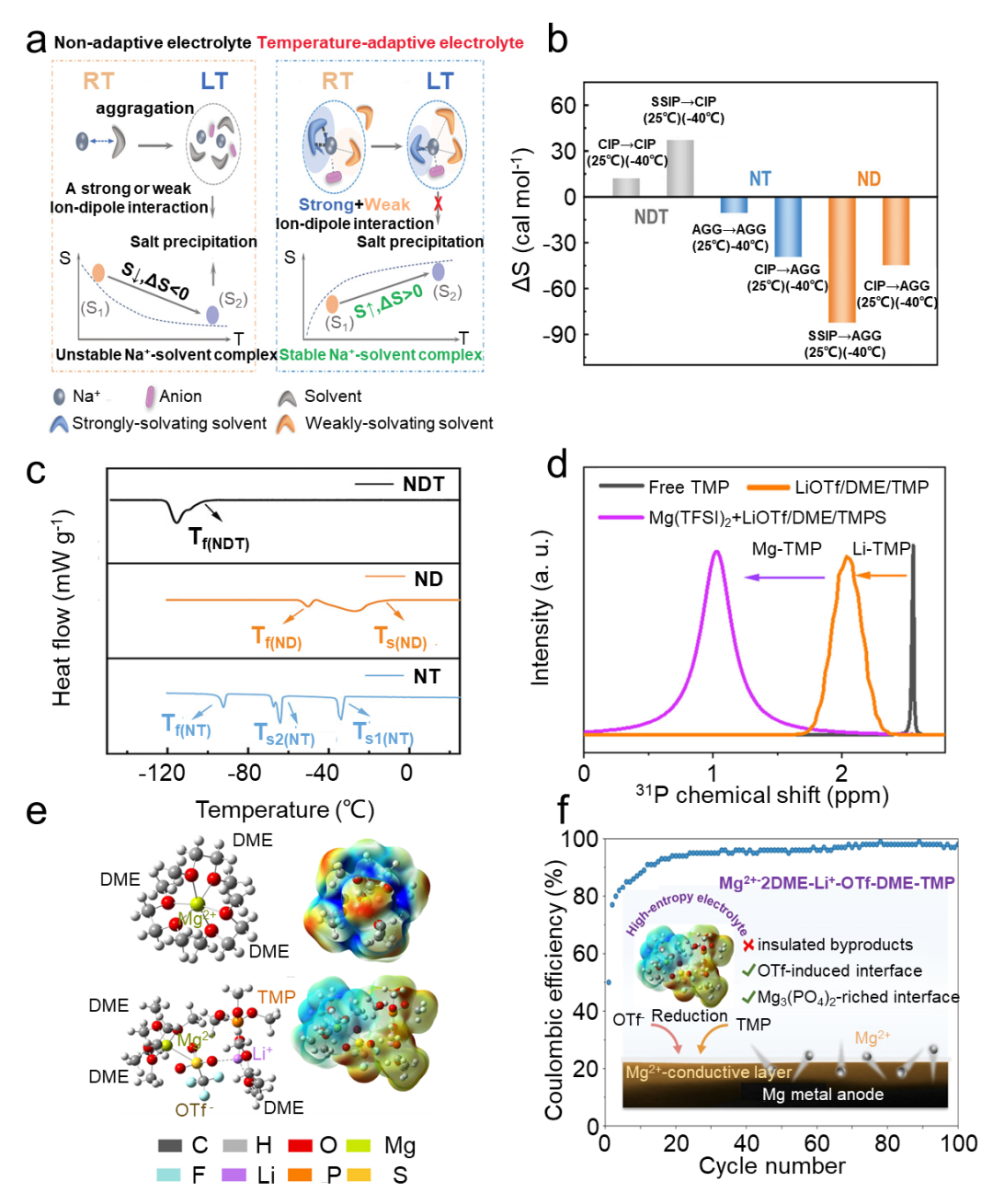
图4 非锂电池用非水液体高熵电解质。(a)低温钠离子电解质设计策略示意图。RT:室温;LT:低温。(b) Na离子电解质溶剂化结构熵变计算值。(c) 各种电解质溶液的差示扫描量热 (DSC) 曲线,其中 Tf 代表凝固温度,Ts 代表盐沉淀温度。(d) TMP溶剂和TMP基电解质的31P NMR谱。(e)Mg(TFSI)2/DME 和 Mg(TFSI)2/DME 与 LiOTf/TMP 电解质的典型溶剂化鞘层和相应的静电电位分布。(f) HEE 衍生的界面反应层(插图)和 Mg||Cu 电池的 CE 示意图。
4. 水性液体高熵电解质
水电解质本质上是安全的、具有成本效益的、低毒或无毒的,因此被认为是大规模储能的合适选择。与有机电解质类似,增加盐的多样性来设计水性 HEE 可以提高盐的溶解度。具体来说,对于水系碱金属离子电池系统,高熵电解质设计可以有效减少游离水量并拓宽稳定性窗口。徐等人。发现不易燃的尿素(CO(NH2)2)-LiTFSI-KOH-H2O三元电解质具有实现高离子电导率和低粘度的潜力,摩尔三角相图如图5a所示。共晶体系中,CO(NH2)2 和 H2O 之间的氢键强度强于 H2O 分子之间的氢键强度,表明 CO(NH 2)2 的添加可以最大限度地减少负极上的界面 H2O 并抑制 H2O 活性,从而将电化学稳定性窗口扩大到>3.3 V(图5b)。另外,添加少量KOH来催化TFSI-的还原。在碱性条件下,F-在高电位下与Li+快速沉淀形成LiF内层,而CO(NH2)2在低电压下在LiF外表面电化学聚合成聚脲。这种双层SEI有利于电化学稳定性。
结果,所制备的Li1.5Mn2O4||Li4Ti5O12软包电池在面积容量为2.5 mAh cm-2 的情况下实现了99.96%的高平均库伦效率,并在470次循环后表现出92%的容量保持率。考虑到电压、倍率性能、环境友好性和循环寿命方面的优点,这种特殊的化学物质有可能超越当前的商业水性技术,例如铅酸、镍镉和镍氢电池(图5c)。水系钠离子电池比水系锂离子电池具有成本优势。郑等人。报道了通过仔细选择钠盐阴离子和优化共晶系统,室温水合物会熔化。在水性 HEE 中,(全氟乙基磺酰基)(三氟甲基磺酰基)酰亚胺钠(NaPTFSI)、双(三氟甲基磺酰基)酰亚胺钠(NaTFSI)和三氟甲磺酸钠(NaOTf)被采用作为前体盐。当共晶成分确定为Na(PTFSI)0.6(TFSI)0.14(OTf)0.21(图5d)时,溶液呈现出最高的混溶性,且含水量极低(H2O/Na+=3.0),相当于18.5M。通过引入具有优异水溶性的稳定的不对称阴离子(如PTFSI-),Na金属水合物熔体不受脆弱的S-F键的影响。值得注意的是,Na水合物熔体电化学窗口(2.7 V)的扩展主要归因于负极极限的扩展。Na水合物熔融电解质氧化稳定性的增强可归因于Na+和水分子之间的强配位,这显着改变了水分子的电子状态(图5e)。Na3V2(PO4)2F3|Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O水合物熔体|NaTi2(PO4)3全电池的平均放电电压为1.53 V,明显高于使用浓NaOTf的电池或 NaClO4 电解质。该电池表现出良好的循环稳定性,在5C倍率下循环500次后仍保持71.6%的容量,平均CE为99.2%(图5f)。
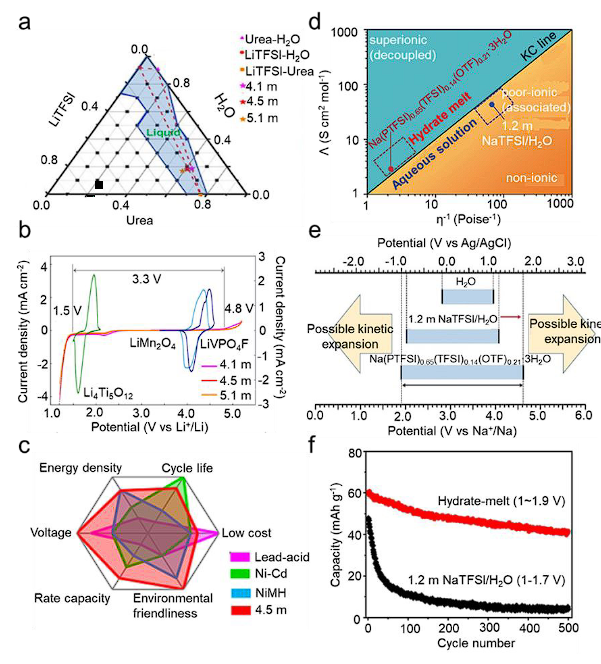
图5 用于水性碱金属离子电池的水性液体高熵电解质。(a) 室温下尿素、LiTFSI 和 H2O 三元相图中的液体区域。(b)具有不同LiTFSI浓度的电解质的总体电化学稳定性窗口,以及集流体上覆盖有循环伏安图的电极的氧化还原。(c) 4.5 m 电解质基水基锂离子电池与其他商业化水基电化学储能技术的比较。(d) Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O 和 1.2 m NaTFSI/H2O 电解质在 25 oC 时的 Walden 图。(e) 纯水、1.2 m NaTFSI/H2O 溶液和 Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O 水合物熔体的电势窗口比较。(f)Na3V2(PO4)2F3|NaTi2(PO4)3全电池在Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O水合物熔体和1.2 m NaTFSI/H2O溶液中的电化学性能。
水性液体 HEE 也已应用于多价离子电池领域,尤其是最有前途的锌离子电池。一般来说,具有高极性溶剂的稀释电解质往往会溶剂化所有离子(如图左下图所示) 6a).溶剂分子之间的强偶极相互作用促进结构有序化,从而导致高玻璃化转变温度 (Tg)。将低熔点非极性或低极性溶剂引入稀释电解质中可以有效降低其粘度,即使在低温下也是如此。然而,这种方法会导致介电常数和载流子浓度降低,最终降低离子电导率。向“盐包水”(WIS) 状态的转变会破坏游离溶剂簇内的分子间网络(图 6a 的右下图)。为了研究局部溶剂化结构的重排,作者分析了水和阳离子的活度系数,并将其与 22°C (2.69 kPa) 纯水的饱和蒸气压进行比较。值得注意的是,Li2ZnCl4 · RH2O (R ≤ 9) 中的水表现出比高水合体系(如 LiCl-H2O 和 ZnCl2-H2O)低得多的活度系数。随着水摩尔浓度从95 mol%下降到67 mol%,对应于盐浓度从1.0 mol kg-1 增加到18.5 mol kg−1 LiTFSI到ZnCl4·6H2O,水的蒸气压从2.33 kPa下降由于Li+离子的充分水合,水活度系数从0.92降至0.088 kPa。这种显着降低的水活度有效地抑制了Li2ZnCl4·RH2O(R≤9)电解质的冻结或沸腾。
鉴于上述考虑,Yang 等人。提出了一种 HEE,其中氯化锂(LiCl)作为支持盐加入氯化锌(ZnCl2)中,形成具有不同 R 的 Li2ZnCl4·RH2O 水性电解质。由于大的体积,Li+会从 ZnCl42- 络合物中解离。水合Li+-ZnCl42-阴离子对的半径(图6a的中图)。同时,Li+离子对的抑制意味着即使在高度浓缩的状态下,它也大部分保持水合状态,这有助于破坏水的氢键网络。由于这种独特的溶剂化结构,Li2ZnCl4的离子电导率9H2O从+80°C时的200 mS cm−1下降到-80°C时的0.66 mS cm−1,超过了为低温操作开发的其他电解质(图6b)。这种卓越的导电性能使其成为低温应用的绝佳选择。由于水活度进一步降低,Li2ZnCl4·9H2O 在-70 °C 下的平均镀锌/剥离CE 可以达到~100%。即使在-80 °C的极低温度下,Zn|Li2ZnCl4·9H2O|ZnxVOPO4·2H2O电池也表现出令人印象深刻的放电容量保持率,即20 °C时容量的81.1%。
最近,邱等人通过考虑离子特异性效应,定量地建立了水分子的熵贡献和冻结温度(Tf)之间的关系,在此期间,四种具有不同阴离子的盐(ZnSO4,ZnCl2,ZnBr2 和 Zn(ClO4)2)被众所周知,单个水分子通常通过氢键与周围的四个水分子相互作用,形成类似四面体的结构。水的 O-H 伸缩振动模式可以解卷积为三个高斯分量,以获得有关不同电解质中氢键 (HB) 的结构和分布的更详细信息。如图6c所示,ClO4- 浓度的增加导致强HB的比例下降和非HB的增加,而SO42-电解质浓度的变化显示非HB的增加可以忽略不计。由此可见,破坏HB的能力顺序为:ClO4->Br->Cl->SO42-。引入四面体序参数(Qtet)的定义来描述局部水四面体度:
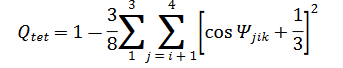
其中 Ψjik 是分子 k 与其最近邻居 i 和 j 形成的角度。一般来说,水分子的Qtet分布在0和1之间(图6d)。Qtet值大于0.8代表冰状水,而该数字低于0.8则代表液态水。如图6d所示,四种不同电解液中的类冰水含量按ClO4-
其中S0是常数,kB是玻尔兹曼常数,P(Qtet)表示给定温度T下Qtet的分布。SQtet的大小受水分子的动力学和热力学性质的影响。在动力学方面,更高的自扩散系数和更短的取向弛豫时间具有提供更多微观状态的能力,有助于更高的SQtet。
从热力学角度来看,S Qtet 较高的水分子表现出较低的 Tf,这可以通过调节阴离子来实现。图 6e 描述了四种电解质中水分子的 SQtet。研究结果表明,当ClO4-存在时,水分子获得了最高的SQtet。得益于水分子与ClO 4- 之间的弱阳离子相互作用以及高SQtet,5m Zn(ClO4)2 电解液在-80 ℃超低温下实现了足够的离子电导率(1.12mS cm-1)。组装好的 Zn|5m Zn(ClO4)2|聚苯胺 (PANI) 全电池在 1 A g-1 下保持了 74.17 mAh g-1 的高容量(图 6f)。总而言之,水性液体HEE表现出广泛的适应性,使其在极端环境下的高安全储能方面非常有前景。尽管如此,水性液体HEE的长期稳定性和耐久性仍有待全面验证。
图6 用于水系锌离子电池的水系液体高熵电解质。(a) 不同电解质中溶液结构的图示。(b) Li2ZnCl4·9H2O 电解质与浓 LiCl·3H2O、ZnCl·3H2O 溶液以及稀(2 mol kg-1)LiCl 和 ZnCl2 水溶液相比的总体离子电导率的阿伦尼乌斯图。(c) 不同浓度的四种电解质中强HB水的比例。(d) 25 °C 下不同电解质中水分子的四面体有序参数 Qtet 的概率分布。插图显示了液态水和冰状水的示意图模型。(e)四种电解质中水分子的四面体熵,在-80至40°C的温度范围内遵循SO42-
5、高熵聚合物电解质
高熵聚合物电解质通常提供增强的电化学稳定性窗口和/或离子传导特性。本节讨论了全固态和准固态高熵聚合物电解质两种高熵聚合物电解质,并详细分析了其特性和电化学性能。
5.1 高熵全固态聚合物电解质
全固体聚合物电解质(SPE)通常由聚合物基质和锂盐作为溶质组成,无需额外的液体增塑剂。高安全性和灵活性的优点使其成为实现全固体电池的重要电解质系统。非晶相和结晶相通常在 SPE 中共存;据信离子传导主要发生在非晶相中。一般来说,对聚合物基体(例如共混、共聚、接枝)、盐和无机填料的高熵改性策略可以抑制聚合物结晶并增加非晶相比例,从而提高离子电导率。对于具有高熵的 SPE -熵聚合物基体,高熵策略带来的亲石官能团可以有效促进分子链中锂离子的运动,从而提高离子电导率。苏等人。通过原子转移自由基聚合和开环聚合,开发了一种具有丰富亲石官能团的星形三元共聚物,然后将其引入聚环氧乙烷(PEO)中,制备了高熵微域联锁全固态固相萃取(HEMI-ASPE)。四种离子偶极相互作用(即聚丙交酯 (PLA) 中的羰基氧和酯氧与 Li+、甲氧基聚乙二醇 (MeOPEG) 中的醚氧与锂离子以及 PEO 中的醚氧与锂离子)以及两个氢-HEMI-ASPE体系中出现了键合相互作用(即聚苯乙烯(PS)末端的末端羟基和溴原子,以及PEO中聚苯乙烯末端的末端羟基和溴原子),构建了动态聚合物网络在微米和纳米尺度上具有高拓扑熵(图7a)。
图7 高熵全固体聚合物电解质。(a) HEMI-SPE 中相互作用的示意图;(b) HEMI-SPE 和 PEO-SPE 在 30-70 °C 时的离子电导率;(c) 21-β-CD-g-PTFEMA 的示意图;(d) 采用 LMFP 正极和 FMC-SPE 的锂金属软包电池第 1、50 和 100 个循环的充电/放电电压曲线;(e)恒电流间歇滴定技术(GITT)和LEHE电解液的Li+扩散参数;(f) 采用 CsPbI3 PQD 的 LEHE 电解质示意图。
在SPE中引入无机填料可以抑制聚合物链的结晶度并增加非晶区,从而提高离子电导率。然而,传统无机填料(例如SiO2 、Al2O3 )与聚合物基体的相互作用不足,限制了电导率增强效果。相反,高熵无机填料可以有效解决这个问题。刘等人。设计了一种具有 Li0.25Mg0.15Co0.15Ni0.15Cu0.15Zn0.15O1-x 配置的 Li 掺杂高熵氧化物(Li0.25HEO)陶瓷,并将其用作 PEO 基 SPE 的填料。71 这种高熵氧化物陶瓷具有岩盐结构不仅表现出高介电常数,而且具有大量的表面氧空位,通过静电引力增强了与 TFSI - 阴离子的相互作用;这些有效地促进了锂盐的解离。
5.2 高熵凝胶聚合物电解质
作为液体电解质和SPE之间的权衡,准固体聚合物电解质(即凝胶聚合物电解质(GPE))受到了研究人员的青睐。高熵 GPE 是指在聚合物基质上具有三个以上重复单元的多组分准固体电解质体系,或采用高熵液体电解质作为增塑剂相。在高熵 GPE 中,聚合物基质提供机械支撑和保持整体结构稳定。
图8 具有高熵聚合物基质的高熵凝胶聚合物电解质。(a) HMPE 制备示意图。(b) Li|1 M LiTFSI 在 1,2-二氧戊环中 0.5 C 下的循环性能:DME 液体电解质@GF 膜|CC-I@GO 和 Li|HMPE|CC-I@GO 电池。(c) NGPE 可能的阻燃机制。(d) DEE(上图)、DEE 与 SFE 的混合物(中图)以及相应的 NGPE(下图)的燃烧测试。(e) NCM811 正极|电解质界面和 Al 集流体在 1 M LiPF-EC: DMC、1 M LiTFSI-DEE 电解质和 NGPE 电解质中的腐蚀行为示意图。
具有高熵聚合物基质的 GPE。
杨等人采用静电纺丝法合成了八(3-氯丙基)多面体低聚倍半硅氧烷(OCP-POSS)改性聚偏氟乙烯/聚丙烯腈/聚甲基丙烯酸甲酯(PVDF/PAN/PMMA)纤维膜(TP- POSS)。将纤维膜浸入 EC/DEC/DMC 液体电解质中的 1 M LiPF6 中后,所得 GPE 在室温下表现出 9.23 mS cm-1 的高离子电导率,并且电化学稳定性窗口具有相对于 Li+/Li 已扩大至 5.82 V。同时,笼状结构 OCP-POSS 的引入以及随之而来的纤维直径的增加增强了 GPE 的机械性能。TP-10%POSS的拉伸强度和断裂伸长率分别为4.70 MPa和74.3%,远高于TP-0%POSS。对于具有增强聚合物相的 GPE,可以通过调整组分之间的相互作用来增加混合熵。增加混合熵可以改善 GPE 的机械性能。他等人。制备了具有夹层结构的高熵带状电解质(HETE)。在该系统中,高熵表层由完全无定形的 PEO-Li+ 复合物组成,有利于快速 Li+ 传输并提供强粘附力。中间层采用高混合熵的PVDF/Li+/PEO,通过Li+增强PVDF和PEO之间的增容作用,提高了电解质的机械性能。具有高熵骨架的HETE表现出优异的机械性能(强度为8.18±0.28 MPa,断裂应变为186.0±12.8%)。
图9 具有高熵液体增塑剂相的高熵凝胶聚合物电解质。(a) 具有离子导电聚酯网络的不可燃 GPE 的合成示意图。(b) 液体电解质的离子电导率和粘度与 TMP 含量的关系。(c) 1 M LiPF6-EC 中 Li+ 沉积过程示意图:EMC 电解质(左)和 HGPE(右)。(d) 1M LiPF6-EC:EMC 电解质和 HGPE 在扫描速率为 5mV s−1 时的线性扫描伏安法 (LSV) 曲线。(e)用于合成高熵水凝胶电解质的组合物的化学结构。(f) 固体聚合物和 12m SAPE的振荡剪切流变学.
含有高熵非水液体增塑剂的 GPE
液体增塑剂相的高熵工程是 GPE 改性的另一种有效方法。交联的凝胶聚合物基质通常含有大量间隙和孔,阻碍离子传输并降低电解质的离子电导率。与聚合物基体具有良好相容性的高熵增塑剂可以很好地填充这些空隙,增强GPE的致密性,提高电极/电解质的接触面积,提高电池性能。帕克等人。通过热固化,开发了一种具有多溶剂的不可燃高熵 GPE。具体来说,使用三种不同的碳酸酯基溶剂(EC、PC、FEC)与 TMP 结合来溶解双(氟磺酰基)亚胺钠(NaFSI)盐,聚己内酯三丙烯酸酯(PCL-TA)用作交联剂(图9a)。当电解质体系达到热力学平衡时,形成4溶剂-Na+构型。合成的GPE具有三维网络结构,不仅可以均匀Na+通量,而且可以封装有机溶剂,有效抑制电极上Na枝晶的生长和溶剂分解。同时,Na+有机溶剂和Na+-聚合物之间的离子偶极相互作用促进NaFSI盐解离成自由离子。优化后的 TMP 含量为 15 vol.%,GPE 表现出不可燃性、非流动性和 6.3 mS cm−1 的高室温离子电导率(图 9b)。合成了高熵全氟化 GPE,其中将 1M LiPF6 溶解在 FEC、2,2,2-三氟乙基甲基碳酸酯 (FEMC) 和 1,1,2,3,3,3六氟丙基-2,2 的混合物中,2-三氟乙醚(HTE),体积比为13。
含有高熵水性液体增塑剂的 GPE。
高熵水凝胶电解质在高性能水系电池中也受到了极大的关注。张等人。报道了一种高熵水凝胶电解质,其合成过程如图9e所示。通过将不同的盐(LiTFSI、LiOTf、N-丙基N-甲基吡咯烷鎓双(三氟甲磺酰基)亚胺(Pyr13TFSI))与水一起溶解来制备前体溶液,单体。在此过程中,LiTFSI 和 LiOTf 用于促进疏水性富含 LiF 的 SEI 的形成,该 SEI 充当电极表面的保护屏障。掺入非挥发性且不易燃的 Pyr13TFSI,以进一步降低水活度并改变锂离子周围的溶剂化结构。基于双酚A乙氧基化物二甲基丙烯酸酯(BEMA)的双官能低聚物被用作交联剂,而聚(乙二醇)甲醚甲基丙烯酸酯(PEGMA)被用作反应性稀释剂,以增强聚合物基质中锂离子的迁移率。紫外光(波长>400 nm)引发的凝胶化大大提高了12m LiTFSI-LiOTf-Pyr13TFSI固态水性聚合物电解质(SAPE)的稳定性和性能。机械性能结果如图9f所示。SAPE 和固体聚合物的弹性模量 (G') 均远高于粘性模量 (G'')。
6.1 高熵氧化物无机电解质
尽管氧化物无机电解质具有较高的化学/电化学稳定性,但其室温离子电导率不足(即10 -4 S cm-1,比商业液体电解质低两个数量级)限制了其应用可行性。高熵可以使氧化物无机电解质的离子电导率提高几个数量级,从而降低体电阻,提高电池的电化学循环性能。曾等人。证明高熵工程引入的化学无序和扭曲会扰乱并重新分布局部位点能量。当这种分布足够宽以实现相邻位点的能量重叠时,就会促进Li+跳跃(图10a)。位点能量的重叠提供了一条扩散路径,沿该路径位点能量变化最小(图10b)。因此,化学无序通过创建能量差异较小的位点的渗滤网络,导致锂离子扩散活化能急剧降低。图10c描绘了在Li-Na超离子导体(NASICON)、NaNASICON和锂石榴石电解质中存在不同掺杂金属种类时计算的位点能量差。这些金属带来的高熵氧化物无机电解质的局部无序通过重叠的位点能量分布有效地促进了位点渗滤并增强了离子电导率。根据计算结果,成功合成了一种新型高熵氧化物无机电解质NASICON Na3.5Mg0.1Sc0.15In0.15Ti0.3Hf0.3ZrSi2PO12,其总电导率高达1.1 mS cm-1,室温下的体电导率为 3.3 mS cm-1。Fu等人在Li6.5La3Zr1.5Ta0.5O12的Zr位点掺杂Nb和Hf元素,Kuo等人在Zr位点掺杂Nb、Y和W元素。离子电导率显着增强,进一步验证了氧化物无机电解质高熵策略的有效性。
图10 高熵氧化物固体电解质。(a) 示意图显示局部扭曲如何造成重叠的站点能量分布;(b) 渗流示意图,定义为最大最近邻站点能量差的站点网络。其中,白球代表渗滤网络外部的能量站点,而绿球代表渗滤网络中具有相似能量的最近邻站点。(c) 计算出 Li-NASICON、Na-NASICON 和 Li-石榴石电解质中各种金属种类的位点能量差异。(d) 立方相和四方相之间的形成焓相对于掺杂剂种类的数量的差异;(e) 通过 DFT 计算获得的振动熵的能量贡献差异。
除了提高离子电导率外,还应用高熵策略来降低立方相石榴石的烧结温度。通过在 Zr 位点使用四种以上的掺杂剂 (Zr-Hf-Sn-Sc-Ta) 合成了三种类型的立方相石榴石,而不形成任何额外的锂空位。熵驱动的稳定效应显着降低了立方相的成核温度。750至400℃发生固体反应。为了解释这种现象,基于密度泛函理论(DFT)计算,根据Zr位点中的掺杂剂数量,比较了四方相和立方相之间的焓差,证明四方相在所有方面都比立方相更稳定。成分(图10d)。这表明,虽然在Zr位点中掺入多种掺杂剂可以减轻内应变,但不足以稳定石榴石立方相的焓形成能,表明具有多种掺杂剂的石榴石中的立方相稳定性可能是稳定的。是由熵效应而不是焓效应引起的。因此,立方相稳定是由电子熵或电子构型熵的增加引起的(图10e)。值得注意的是,稳定的四方相在低温下逐渐形成,其稳定性随着掺杂金属元素的增加而提高。高熵Li6.6La3Zr0.4Hf0.4Sn0.4Sc0.2Ta0.6O12的体积离子电导率在25℃时为3.2×10-4 S cm-1,而Li|Li7La3Zr0.4Hf0.4Sn0.4Sc0.4Ta0.4O12 |LiNi1/3Co1/3Mn1/3O2 电池表现出稳定的循环性能,在 700 次循环后仍保持超过 143 mAh g-1 的高容量,容量保持率为 92%。总之,高熵策略可以有效提高氧化物固体电解质的锂离子电导率并降低其烧结温度,这为氧化物电解质的发展提供了新的见解。然而,高熵氧化物电解质中使用的贵金属增加了成本。因此,筛选具有成本效益的高熵氧化物电解质替代金属仍然是一项具有挑战性的任务。
6.2 高熵硫化物/卤化物无机电解质
在全固体电解质中,硫化物/卤化物无机电解质具有最高的离子电导率,在没有任何掺杂剂的情况下达到10-3 S cm-1。对于硫化物/卤化物无机电解质体系,高熵策略可用于进一步提高其离子电导率,与商业液体电解质相当(即室温下 10-2 S cm-1)。混合聚阴离子理论是一种指导高熵硫化物无机电解质设计的有力方法。据信,晶体结构中各种聚阴离子基团的存在通过降低锂离子迁移的能垒来提高阳离子迁移率。在Li2S-GeS2-LiBr-LiI体系中观察到一个典型现象,其中S2-、I-和Br-阴离子的混合降低了活化能并提高了阳离子迁移率。李等人。报道了一种高熵硫化物电解质,即 Li9.54[Si0.6Ge0.4]1.74P1.44S11.1Br0.3O0.6.图 11a 显示了两个成分参数的图,即晶体结构指标 (t) 和组合复杂度度量(Smix)。前者是组成阴离子(Vsum-anion)与阳离子(Vsum-cation)的总体积之比,可以根据离子半径计算。后者可以根据阴离子和阳离子种类的占据紊乱来计算。据此,根据设计指南合成了Li9.54[Si0.6Ge0.4]1.74P1.44S11.1Br0.3O0.6,以追求高Smix值和可行的t值。所开发的硫化物 HEE 的室温体积电导率为 32 mS cm-1,大约是原始 LGPS 的三倍(图 11b)。这些极高的电导率解释了在上述固态电池(SSB)中使用毫米级厚正极的可行性。
图11 高混合熵硫化物无机电解质。(a) 报告的 LGPS 型(空心圆圈)和银汞矿型(空心方块)电解质的晶体结构指标 (t) 和成分复杂性度量 (Smix) 之间的关系;(b) Li9.54[Si0.6Ge0.4]1.74P1.44S11.1Br0.3O0.6 和 LGPS 的体电导率的阿伦尼乌斯图;(c) 锂金属电池在25℃至-10℃不同温度下0.025C下的放电曲线;(d) 电流密度在 0.2 和 0.68 mA cm-2 之间的现有和报道的硫化物电解质电池在 25 °C 时的面积容量比较;(e) 25℃及更低温度下的面积容量(下)和放电容量保持率(上)的比较。
如上所述,银汞矿结构中的阴离子亚晶格通常在威科夫4a和4d位置处的二价S2-和一价卤化物(Br-或Cl-)之间表现出位点无序。因此,为了研究阴离子混合熵和Li+传导之间的影响,Wang等人合成了一系列富含卤素的银汞矿 Li5.3PS4.3Cl1.7xBrx (0≤x≤1.7)。阴离子结构框架的灵活性为可调离子电导率提供了新的化学。Li 5.3PS4.3ClBr0.7 的晶体结构如图 12a 所示,其中硫和卤素阴离子的不稳定交换发生在 Wyckoff 4a 和 4d 位点上。阴离子混合影响邻近锂离子的位点占用,促进笼间跳跃以实现远距离Li + 扩散并提高离子电导率。计算结果还表明,在Li5.3PS4.3Cl1.7-xBrx系列的中间,4d位点的S、Cl和Br的分数接近其组成指标。(图12b)。采用最大熵法(MEM)分析来检查溴化对 PS43− 重新取向的影响。可以观察到,时间平均 S 核密度相对集中在 Li6PS5Cl 内的原始位置,表明 PS43− 的运动有限。
此外,硫化物是具有高离子电导率的陶瓷,同样可以通过应用高熵来设计合金策略。赵等人。开发了一种二维(2D)高熵过渡金属磷硫化物,即 Lix(Fe1/5Co1/5Ni1/5Mn1/5Zn1/5)PS3 (HE-LixMPS3),作为锂离子导体。当过渡金属元素数量达到5时,LixMPS3相的吉布斯自由能下降至-0.83 eV/晶胞,表明高熵LixMPS3由于其负自由能更大,比过渡金属元素较少的对比样品更稳定(图12e)。然而,Li xMPS3 随着 ΔG 增加过渡金属元素数量超过 5 的变化仍然值得怀疑,因为已证明辛元石榴石和二元石榴石固体电解质的相稳定性和离子扩散较差于五元石榴石高熵效应产生的高晶格畸变和大量金属阳离子空位为 Li+传输到 HE-LixMPS3 结构中构建了高速公路,同时 HE-LixMPS3 的二维层间纳米通道有利于锂离子。由于这种高熵设计,HE-LixMPS3在室温下的离子电导率为5×10-4 S cm-1,远高于其他未经高熵改性的电解质样品(即10-5-10-8 S cm-1)。总而言之,高熵策略对于无机电解质至关重要。更多的高熵掺杂机制仍有待开发和应用,而在非锂电池系统中的应用应进一步扩大和实用化。
图12 高构型熵硫化物/卤化物无机电解质。(a) Li5.3PS4.3ClBr0.7 的晶体结构,由 Rietveld 精修获得。(b) Li5.3PS4.3Cl1.7−xBrx中Cl(青色,实心正方形)、Br(橙色,实心菱形)和 S(蓝色,空心正方形)分别占据 Wyckoff 4d 位点的分数。(c) Li 5.5PS4.5ClxBr1.5-x.中 Sconf 与离子电导率之间的相关性。(d) 不同 UCl3 型氯化物电解质的阿伦尼乌斯电导率图。(e) 具有不同金属成分(M = Fe、Co、Ni、Mn、Zn)的 LixMPS3 相在 1023 K的示意图和吉布斯自由能
总结与展望
HEE 代表了一类新型电解质,具有许多独特的特性。它们的突破性发展将彻底改变液体、准固体和全固体可充电电池,不仅推动电解质设计理论的进步,而且有助于弥合实验室研究和生产应用之间的差距。在这篇综述中,我们总结了不同种类 HEE 的性能特征,包括离子电导率、热/化学/电化学稳定性以及与电极材料的兼容性。此外,还回顾了 HEE 在各种可充电电池中的应用,以解决其关键缺点(例如低温性能不足和循环寿命有限)。尽管潜力巨大,但高能电子的探索仍面临以下挑战:
图13 可充电电池用HEE的发展和前景
文献链接:
X. Zhao, Z. Fu, X. Zhang, X. Wang, B. Li, D. Zhou and F. Kang, Energy Environ. Sci., 2024, DOI: 10.1039/D3EE03821A.
原文链接:
https://pubs.rsc.org/en/content/articlelanding/2024/ee/d3ee03821a
作者介绍
康飞宇教授 清华大学深圳国际研究生院教授、博士生导师。入选“广东特支计划”和深圳市杰出人才,2018-2021年连续入选科睿唯安“全球高被引科学家”,以第一完成人获国家技术发明二等奖、教育部自然科学一等奖、广东省自然科学一等奖和中国建材学会技术发明一等奖,并获深圳市长奖和广东省丁颖科技奖。作为材料科学与技术专家,在储能用碳材料和先进电池方面取得系统性创新成果。2012年发现并阐明了锌离子可逆电化学反应机制,建立了多价离子存储理论(Angew. Chem. Int. Ed., 2012, 51, 933),有力地推进了水系二次电池的产业化进程。作为首席科学家主持了国家重大科学研究计划项目和多项国家自然科学基金重点项目、科技部863项目和科技攻关计划。迄今发表SCI论文450余篇,他引35000余次,60篇曾入选ESI高被引论文,H因子108,合著中英文著作8部,授权发明专利160件(包括美日韩专利12件),31件实现技术转移和应用。
周栋助理教授 周栋博士现为清华大学深圳国际研究生院助理教授、博士生导师。2017年博士毕业于清华大学材料学院,师从康飞宇教授。其后先后加入澳大利亚悉尼科技大学清洁能源技术中心和日本东京大学理学院从事博士后研究。研究方向主要为用于新型二次电池体系的关键电解质材料和电极材料的设计与制备;先进二次电池体系中电极|电解质微观界面电化学的研究;以及新型纳米/聚合物材料在能源、环境科学领域的应用及机理探究等。目前以第一/通讯作者在Nat. Energy, Nat. Nanotechnol., Nat. Commun.(4), Nat. Rev. Mater., Chem. Rev., Chem (2), J. Am. Chem. Soc. (2), Angew. Chem. Int. Ed. (7), Adv. Mater.(3), Energy Environ. Sci. (2), Nano Lett.(2)等期刊发表论文40余篇,总被引七千余次(谷歌学术)。获得国家高层次青年人才资助,当选日本JSPS Fellow、澳大利亚DECRA Fellow。2020年获得广东省自然科学一等奖。
审核编辑:刘清
除了星形三元共聚物链段的功能外,HEMI-ASPE 的超分子相互作用赋予了优异的机械性能,保护系统中暴露的羟基。HEMI-ASPE的互穿网络限制了半径较大的阴离子的运动,从而将70℃下的离子电导率提高至4.56×10-4 S cm -1,是基于PEO的SPE的两倍多(图1)。.7b).与这项研究类似,Su 等人。设计了一种含氟聚合物 (21-β-CD-g-PTFEMA),具有 β-CD(β 环糊精)和聚(2,2,2-三氟乙基甲基丙烯酸酯)(PTFEMA)(图 7c)。通过将所开发的高熵21-β-CD-g-PTFEMA引入PEO中,制备出具有高迁移数(tLi+ = 0.88)的富氟高分子全固体SPE(FMCASPE)(图7c) )。FMC-ASPE中的三个极性基团(即C=O·Li +、C-O·Li + 和CF·Li+)可以协同改善锂离子的运动,赋予更高的离子电导率和传输数。
此外,两种弱氢键(即O-H·F-C和C-H·F-C)可以有效降低结晶度,提高机械强度。此外,它们还可以保护PEO末端羟基免于氧化/还原,阻碍TFSI−阴离子激发,并增强富氟FMC-ASPE的热稳定性。结果,高电压 LiMn0.6Fe0.4PO4 (LMFP)|FMC-ASPE|Li 软包电池在 70 oC 下表现出超过 100 次循环的延长循环寿命(图 7d)。总之,高熵策略可以将SPE的离子电导率提高一个数量级以上,并提高电池的循环稳定性。然而,仍需要付出更多努力,包括合理调整聚合物链段以进一步提高离子电导率并扩展超锂电池的高熵SPE。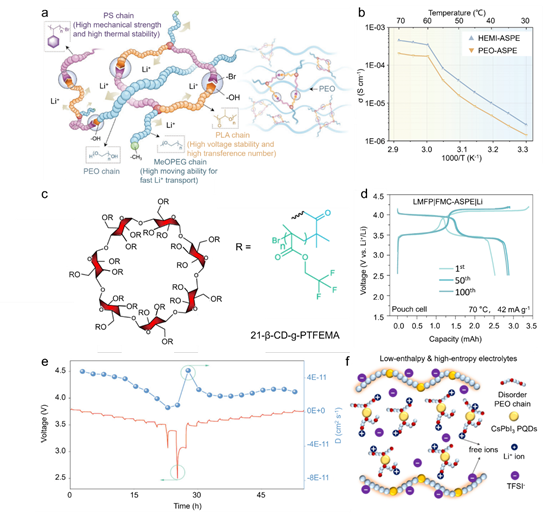
因此,Li0.25HEO填料阻断了PEO链的结晶度,进一步提高了离子电导率。当在 SPE 中引入 15 wt% Li0.25HEO 填料时,PEO-15Li0.25HEO 电解质在室温下提供了令人满意的电导率 8.9 × 10-5 S cm-1 ,比原始 PEO 基 SPE 高两个数量级。这项工作为 SPE 的发展提供了一条新途径,尽管这种高熵 SPE 的离子电导率仍然低于具有超离子导体填料(例如 Li 6.4La3Zr1.4Ta0.6O12)的 SPE。72 张等人。通过将 CsPbI3 钙钛矿量子点 (PQD) 引入 PEO@LiTFSI 复合物中,构建了低焓高熵 (LEHE) SPE。PEO-LiTFSI 电解质的扩散系数从 1.76 × 10-11 cm-2 s-当引入 1.5 wt.% PQDs 填料时,为 1 至 2.75 × 10-11 cm-2 s -1 (图 7e)。因此,在最佳添加量为 1.5 wt.% 的情况下,LEHE 的室温离子电导率最高为 1.4×10-4 S cm-1。优化效果可归因于CsPbI3 PQDs显着抑制PEO链的结晶度,并赋予PEO-LiTFSI-CsPbI3复合物高熵特性(图7f)。进一步研究开发具有高本征锂离子电导率的高熵无机填料有利于 SPE 的进一步优化。
此外,聚合物基体的高熵结构赋予 GPE 多个离子通道,从而促进离子传输。同时,高熵 GPE 中的液体增塑剂可以增强 GPE 的柔韧性和稳定性,有助于承受电池过程中的变形和应力。高熵液体增塑剂还具有增强界面相容性的优点。与 SPE 相比,GPE 通常与电极表现出更好的界面接触/相容性,使其更适合适应现有的 LIB 生产路线。通过共混或共聚设计来设计具有三维网络结构的高熵聚合物基体,高熵 GPE 可以提高机械强度和稳定性。这对于防止 GPE 内变形或断裂至关重要,特别是在电池充电和放电过程中发生的体积变化过程中。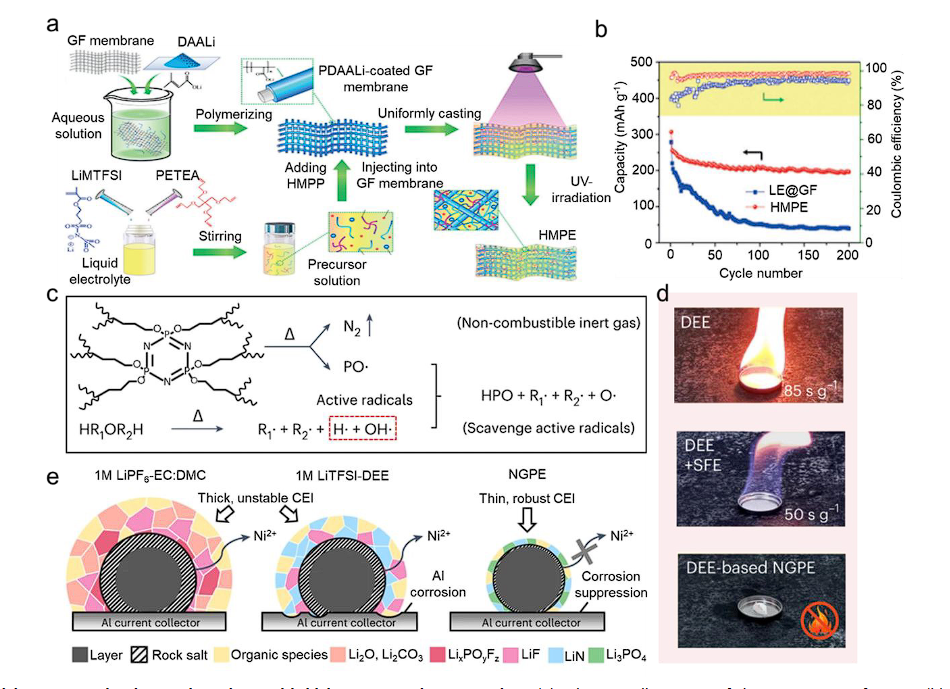
同时,HETE的穿刺强度(66.9±1.4 g/25.4 μm)与高熵骨架的穿刺强度(76.0±1.5 g/25.4 μm)相似,HETE的韧性测得为11.28±1.12 MJ m -3 得益于 PVDF 的高混合度。HETE的带状附着力使得带状电池能够以附着力主导、无压缩的方式设计和组装,促进了结构集成准固态电池的发展。高熵聚合物基体的分级结构设计可以很好地平衡GPE的机械强度和导电性,从而实现优异的电池性能。周等人。报道了一种新型分级多功能聚合物电解质(HMPE),由原位聚合的1-[3-(甲基丙烯酰氧基)丙基磺酰基]-1(三氟甲磺酰基)酰亚胺(LiMTFSI)-季戊四醇四丙烯酸酯(PETEA)基交联凝胶聚合物电解质组成。3,3-二甲基丙烯酸锂(PDAALi)涂层玻璃纤维(GF)膜(图8a)。PDAAL涂层赋予HMPE足够的机械强度(4.7 MPa),而LiMTFSI-PETEA共聚物网络保证了操作安全。由于含有分层单离子导体结构,所制造的 HMPE 表现出所需的离子电导率和 0.75 的高 Li+ 迁移数。Li|HMPE|氧化石墨烯包裹的碳布-碘(CCI@GO)全电池具有出色的循环性能,平均CE为98%(图8b)。
此外,原位凝胶反应中的残留单体可用于实现增塑相和聚合物骨架的双重高熵改性。,Meng等人设计了一种不易燃的基于醚的 GPE (NGPE),以解决安全问题并提高基于醚的电解质的电极兼容性。将氟甲基1,1,1,3,3,3-六氟异丙醚(SFE)引入乙醚(DEE)溶剂中,随后通过丁烯氧基环三磷腈的共聚将二元电解质原位转化为凝胶态(BCPN) 单体和季戊四醇丙烯酸酯 (PETEA) 交联剂。当受热时,PO·自由基从聚合的BCPN中释放出来。这些自由基有效地捕获醚溶剂热分解产生的气态氢自由基和氧自由基,从而阻碍放热链式反应。
此外,聚合BCPN分解过程中产生的惰性气体(例如N2)进一步阻碍了燃烧过程(图8c)。进行分子动力学(MD)模拟以证实电解质基质的溶剂化结构。溶剂化结构中阴离子的参与可能会导致锂金属表面阴离子还原产生坚固的表面膜。这些含有离子锂化合物(例如LiF)的表面薄膜充当固体电解质界面(SEI),保护反应负极免受破坏性界面副反应的影响,从而增强锂金属负极的循环性能。还测量了电解质的溶剂化能(ΔGsolv)以进一步探索溶剂化结构。1 M LiTFSI-DEE:SFE 电解质和 NGPE 表现出比 1 M LiTFSI-DEE 参比电解质 (-153.8 mV) 更大的负开路电位(分别为 -212.7 和 -216.8 mV)。Li离子与DEE分子之间配位的减少是SFE作为反溶剂作用的结果,这与Li离子迁移数的增加密切相关。结果,通过加入 5 wt.% BCPN,NGPE 表现出完全不可燃性,自熄时间为零(SET,图 8d)。组装Li||LiNi0.8Co0.1Mn0.1O2 (NCM811)全电池以进一步证明所制备的NGPE的有效性。与 1 M LiTFSI-DEE 和 1 M LITFSI-DEE:SFE 电解质中形成的厚且不稳定的正极电解质中间相 (CEI) 不同,含有 NGPE 的电池在正极表面上表现出更薄且更优化的 CEI,这是由残留的 BCPN 产生的单体(图8e)。在正极上形成的这种均匀的富含磷和氮的保护表面膜可以有效地抑制有害的副反应和Ni2+ 离子的溶解。结果,使用NGPE的Li||NCM811电池表现出约200mAh g-1的高初始放电容量,并且500次循环后的容量保持率超过75%,平均CE为99.5%。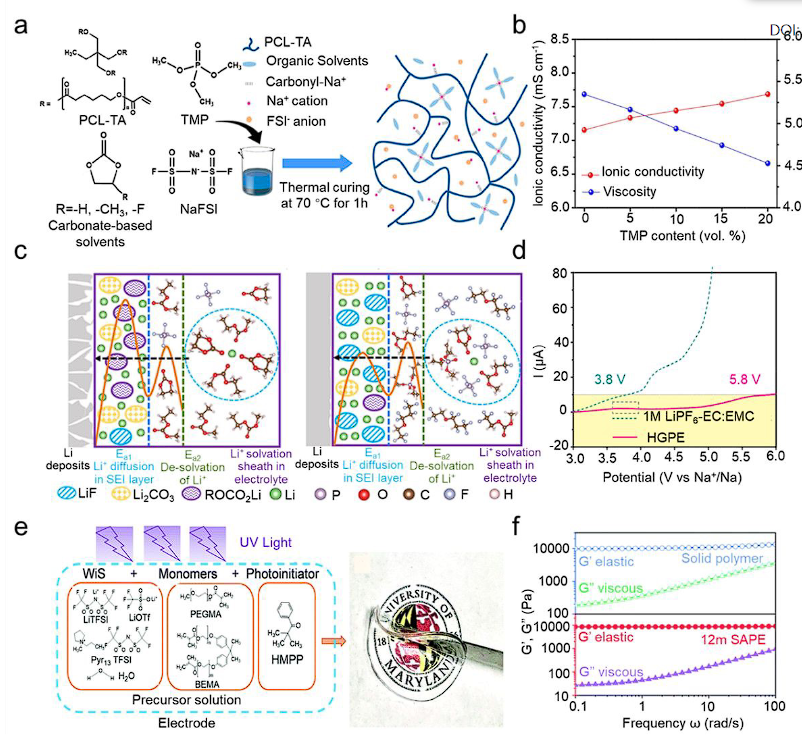
在该系统中,FEC 增强了与 Li 负极的兼容性,而 FEMC 确保 PF6- 可逆地插入/脱出到石墨中。HTE 充当稀释剂,降低电解质粘度并优化阳离子/阴离子聚集体的局部溶剂化结构。此外,在该氟化电解液中添加3重量%二乙基烯丙基磷酸酯(DAP)单体和1.5%季戊四醇四丙烯酸酯(PETEA)交联剂,形成杂原子基GPE(HGPE),通过防止液体泄漏,有效提高电解液的安全性。添加 HTE 作为稀释剂导致在电解质中形成局部高度集中的区域,其中氟化碳酸盐和 Li+-阴离子离子对参与溶剂化壳。这种溶剂化壳结构和 HGPE 中残留的 DAP 单体导致在锂金属表面形成无机组分(例如富含 LiF)SEI,该SEI 在抑制枝晶形成方面非常稳健,并在整个循环过程中保持低电阻。此外,交联的 DAP-PETEA 基质产生均匀的 Li+ 通量,并有效缓解 Li 沉积时的体积变化,从而抑制任何初期枝晶生长(图 9c)。同时,HGPE在5.8 V之前都观察到低氧化电流(图9d),证明了其在高电压锂金属电池中的应用潜力。
此外,G' 与频率无关,而 G'' 随着频率逐渐增加。这表明 SAPE 的行为更像固体电解质而不是液体电解质。12m SAPE 的 G' 值为 8.5 kPa,与固体聚合物基体的 G' 值 (10 kPa) 非常接近。所开发的12m SAPE使得无隔膜的LiMn2O4||Li4Ti5O12水系全电池在0.5 C下具有稳定的能量密度151 Wh kg-1。总之,增塑剂相和聚合物基体之间的协同效应有效地增强了高熵 GPE 的机械和电化学性能。然而,新兴高熵GPE相对复杂且经济上不合理的制备技术仍需要进一步优化。高熵无机电解质与商用非水液体电解质和SPE相比,无机固体电解质由于其固有的不可燃性而具有更好的安全性。当与锂金属负极配合时,基于无机固体电解质的全固态锂金属电池与商用锂离子电池相比,能量密度显着提高。因此,氧化物(例如,Li1.3AI0.3Ti1.7(PO4)3 (LATP)、Li7La3Zr2O12 (LLZO))、硫化物(例如,Li10GeP2S12 (LGPS)、Li6PS5Cl (LPSCl))和卤化物(例如,Li3YCl6、 Li3InCl6)无机电解质近年来引起了越来越多的关注。然而,主流无机固体电解质的室温离子电导率无法与液体电解质相比,这限制了无机电解质的应用。此外,氧化物无机电解质的高合成温度(>700 oC)增加了生产成本。高熵策略是解决这些问题的有效解决方案。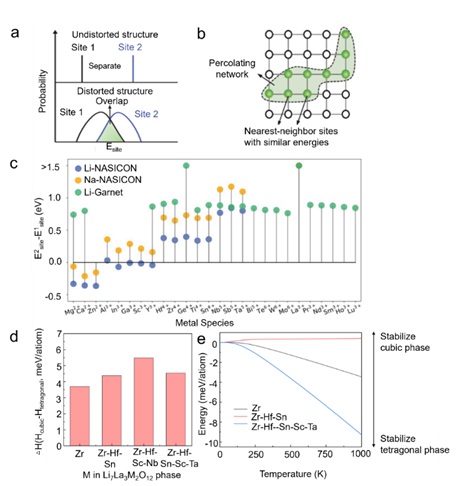
当应用于使用面积负载为 245 mg cm-2 的 LiNbO3 涂层 LiCoO2 正极的锂金属电池时,基于硫化物 HEE 的电池提供了 22.7 mAh cm-2 的面积放电容量,输入利用率为 97% LiCoO2在25°C时的截止电压为4.25 V vs. Li+/Li(图11c),超过了大多数报道的基于硫化物电解质的锂金属电池(图11d)。-10℃时的放电容量仍是25℃时的75%,高于大多数报道的基于硫化物电解质的锂金属电池(图11e)。除了Smix之外,构型熵(即Sconf)也被提出用于高化学无序硫化物/卤化物无机电解质的设计。林等人。开发了 Li6.5[P0.25Si0.25Ge0.25Sb0.25]S5I 固体电解质,以研究 Sconf 对 LGPS 电解质的离子电导率和结构稳定性的影响。S2- 和 I- 均形成面心立方亚晶格。(即威科夫位置 4a 和 4d),而 S2- 位于八面体位点(即威科夫位置 4b 上的中心原子)周围的一半四面体空隙(即威科夫位置 16e)中。这形成了由[PS4]3-、[SiS4]4-、[GeS4]4-和[SbS4]3-组合而成的聚阴离子[P0.25Si0.25Ge0.25Sb0.25S4]3.5-四面体。。考虑到四面体环境中 4b 位点上元素的均匀分布以及上述位点反转(即威科夫位置 4a 和 4d),Sconf 计算为 2.03 R(理想气体常数),表明Li6.5[P0.25Si0.25Ge0.25Sb0.25]S5I固体电解质。高熵聚阴离子结构为固体电解质提供了约0.2 eV的低Li扩散活化能和13 mS cm-1的高室温离子电导率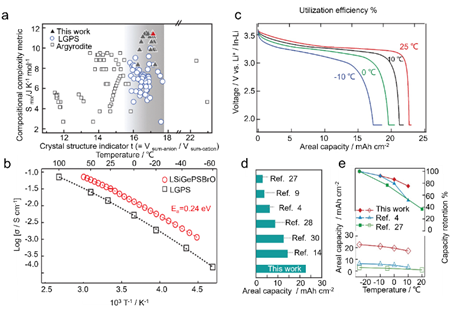
因此,这个最大阴离子熵区域提供了最高的离子电导率;当 x=0.7 (Li5.3PS4.3ClBr0.7) 和 1.0 (Li5.3PS4.3Cl0.7Br) 时,富 Cl 和富 Br 组合物的最高电导率分别为 24 和 26 mS cm-1。Li等人合成了一系列银辉石 Li5.5PS4.5ClxBr1.5-x (0 ≤ x ≤ 1.5) 并计算了它们的构型熵值。如图12c所示,当元素Cl和Br分布更均匀时,构型熵变得更高。高熵设计带来的无序对锂离子的动态产生积极影响,导致Li5.5PS4.5Cl0.8Br0.7的室温离子电导率高达22.7 mS cm-1,ΔSconf= 1.98 R。此外,Li5.5PS4.5Cl0.8Br0.7电解质使单晶LiNi 0.9Co0.06Mn0.04O2复合正极在SSB中具有出色的循环稳定性,即在2 C倍率下循环超过700次而没有明显的容量衰减而且,Sconf的概念还可以用来指导卤化物无机电解质的设计。Li 等人使用多重混合策略。使用具有成本效益的 LaCl3 和 CeCl3 晶格作为主体,设计了一系列 UCl3 型氯化物电解质。UCl3 型结构可以描述为球体的扭曲六方最密堆积,可以推广到 MX3 成分(M = La -Gd,X是卤素)。X占据了Wyckoff位置6h,同时M1原子占据了位于(002)面的Wyckoff 2c位置,也形成了[MX9]6-多面体。这些[MX9]6多面体还共享边缘以在[001]方向形成六角形通道,并且[MX9]6-多面体中的柱的排列在[001]方向提供八面体和三棱柱空位点以用于多个阳离子传导。图 12d 显示了这些多阳离子混合氯化物电解质的离子电导率的阿伦尼乌斯图。值得注意的是,室温下的离子电导率可以提高四个数量级,从 10-7 S cm-1 水平(对于原始 Li-La-Cl 系统)到 10-3 S cm-1 水平(对于多种阳离子配方系统,例如,通过多重阳离子混合策略,LiCl-(LaCl3·CeCl3·ZrCl4·HfCl4·TaCl5)0.2)为1.8×10-3 S cm-1 。因此,组装后的 NCM88 全电池可以稳定循环 3000 次,在 5 mA cm2 的高电流密度下容量保持率为 80%。锂基电池用硫化物/卤化物无机电解质的性能和制备方法总结于表1。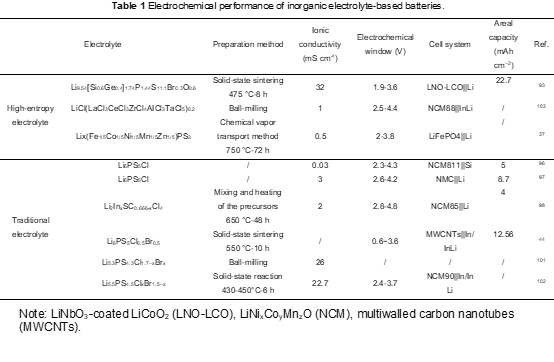
(1)定义模糊。电解质研究中“高熵”的定义仍然存在争议和模糊。(2) 组件优化过程效率低下。目前,筛选适合 HEE 的组件仍然很大程度上依赖于试错法。非常需要超越现有广泛指导方针(例如共晶相图)的更有效的筛选技术。(3)电解质稳定性可疑。由于 HEE 中存在多种组分,因此需要仔细检查其热/化学/电化学稳定性受复杂组分相互作用甚至副反应的影响。(4)幼儿实际应用。目前大部分高能电子研究成果还停留在实验室阶段,需要更多务实的测试和评估。
因此,需要在以下领域做出巨大努力:
(1)由于HEE领域仍在不断发展,正在进行的研究应在深入了解组分比例、相互作用和相的影响的基础上为其发展建立更清晰的定义和指南。HEE 特性的行为。在设计HEE时,需要选择由不同元素组成的化合物,以确保电解质中各种离子的平衡,同时保证成分之间的高相容性和低化学反应性。对于高熵固体电解质来说,至少需要5种不同的元素才能形成稳定的成分结构。对于液体 HEE,溶剂化结构中优选包含多种阳离子、阴离子和/或溶剂的熵有利的小离子簇。虽然多组分混合可以增加电解质体系的无序程度,增强热力学稳定性和抗结晶能力,但仍需进一步筛选所开发的HEE,以确保充放电循环过程中令人满意的电化学稳定性,从而赋予电池耐久性。然而,新兴系统中的“熵”概念仍然含糊不清(例如,Sconf 或 Sex)。理论模拟和建模(例如 MD)有望揭示 HEE 组分的相/结构/性能变化与基本物理化学参数之间的内在联系。对于全固体无机HEE,由于其对离子电导率的关键影响,Sconf的评估应引起足够的重视。仍需要在高熵设计方面做出更多努力,以进一步提高无机 HEE 的离子电导率,使其与液体电解质相当(图13,左上图)。
(2) 在 HEE 中,由于每种附加成分的可能组合数量呈指数增长,因此预测最佳组合是一个难题。相图分析被认为是克服这些问题的一种策略,因为相图提供的成分比例、相稳定性、相变温度等信息可以指导 HEE 成分的筛选。例如,选择共晶或固态成分-溶液特性可以提高液体 HEE 的稳定性和离子传输性能,这有利于稳定性增强和离子传输加速。此外,基于人工智能/机器学习 (AI/ML) 方法的理论计算显示出巨大的潜力在解决筛选新型电解质成分的质量参数和数据挑战方面,同时高通量协同筛选技术可以指导HEE的合成和测试过程,并准确评估目标HEE的可行性。此外,值得注意的是,考虑到 HEE 的实际应用,应考虑其成本。例如,筛选廉价的替代元素来取代大多数新兴全固体无机HEE中昂贵的金属元素,同时降低合成温度对于其SSB的开发具有关键意义(图13,左下图)。
(3) HEE的稳定性直接影响电池的日历寿命。然而,由于传统表征方法的局限性,HEE 与界面相关的热/化学/电化学稳定性仍然难以精确评估。因此,应采用先进的原位/操作(例如,原位固态核磁共振和原位中子技术)和/或无损表征技术(例如,低温透射电子显微镜)来进行实时、动态和直观的研究高熵溶剂化/晶体结构与界面稳定性之间的相互作用。具有互补技术的综合方法可以进一步呈现界面副反应持续形成和演化过程的更全面的图景,并提供结构-性能关系的可靠反馈,以指导高度稳定的HEE的合理设计。还值得注意的是,充电电池的极端工作环境对HEE的设计提出了更高的要求。例如,对于高温应用,优选具有较高熔点和热稳定性的电解质组件,而低温场景则需要具有较低熔点和快速离子传输能力的组件(图13,右上图)。
(4) HEE的制备成本和HEE基电池的生产成本是商业化的重要考虑因素。优化生产工艺、简化材料合成方法、提高生产效率可以增强HEE在锂基电池中的商业竞争力,同时,还需要在HEE向非锂电池系统的应用拓展方面付出更多努力。例如,钠离子和锌离子电池)以及可行性验证。为了创建具有高安全性、高效率和适应性的电池模块,必须彻底评估基于 HEE 的电池在各种操作和老化条件以及极端环境条件下的特性(图 13,右下图)。总之,HEE表现出优异的电化学和热稳定性,使其在高能碱金属(如Li、Na)基电池和多价离子(如Mg、Zn)电池中得到广泛应用,促进了电池的发展设备朝着更轻、更便宜、更可靠的方向发展。对于应用于锂基电池的HEE,提高Sconf是提高电解质离子电导率的有效方法。
由此,可以提高电池动力学和低温容量。对于应用于水性多价电池(例如锌离子电池)的液体HEE,Stet的调节可以有效抑制水反应性并减少析氢反应,从而改善电池循环过程中的CE和容量保持率。对于应用于非水多价离子电池(例如镁离子电池)的液体 HEE,可以通过调节 Smix 来很好地调节电解质离子电导率和界面成膜性能。结果,多价金属负极电镀/剥离的CE和电池寿命可以显着提高。此外,HEE 还可以抑制溶剂的冻结和沸腾,使其在极端温度环境中具有潜在用途,例如极地或外太空的储能设备。考虑到这一有吸引力且重要的领域的快速进展,我们有理由相信高能电子器件将在下一代储能技术中发挥关键作用,为清洁能源和可持续发展做出贡献。
-
不同类型的电池的电解质都是什么?2024-02-27 4313
-
可充电电池和快速独立充电器概述2023-03-23 3450
-
弱溶剂间相互作用提高电池电解质稳定性2023-03-13 3563
-
固态电解质引入特殊官能团实现高电压锂金属固态电池2022-11-16 4596
-
相变电解质助力高稳定性锂金属电池2022-10-25 3414
-
钠离子电池的电解质分类2022-10-09 6766
-
按电池中电解质性质分为哪几种2021-08-31 2239
-
新型固体材料可替代电池中的易燃液体电解质2020-09-25 1446
-
将商业化锂离子电池中的液态电解质替换什么解质?2020-06-09 3500
-
10微米厚的陶瓷电解质 让固态电池充电速度更快2020-03-24 5634
-
水溶液电解质电池的安全使用建议2012-05-30 1694
-
可充电电池2012-05-10 8865
-
电池内的电解质是什么?2009-10-20 1244
-
12V胶状电解质电池充电电路2009-01-10 963
全部0条评论

快来发表一下你的评论吧 !
