

高压、快动力学钠金属电池中的竞争配位
描述
研究背景
钠基电池因其与锂基电池相似的储能机制以及钠资源分布广、成本低等特点,被认为是锂基电池的互补技术。为了实现更高的能量密度,毫无疑问,钠金属是钠基电池的最终负极,因为它具有高理论比容量(1166 mAh g–1 )和低氧化还原电位(−2.714 V vs SHE)。然而,钠金属负极面临的问题与锂金属负极相似甚至更严重。钠金属反应性更高,与电解液发生更严重副反应,形成化学/电化学稳定性较差的脆弱固体电解质界面(SEI)。在随后的重复剥离/电镀过程中,SEI溶解并破裂,进一步导致钠枝晶的生长和死钠的形成。这种不稳定的界面结构导致活性钠的不可逆损失,严重恶化了钠金属电池(SMBs)的安全性和循环稳定性。此外,钠的氧化还原电位 (+0.3 V vs Li) 高于锂,这意味着在全电池相同的充电截止电压下,SMBs 中使用的电解液需要更高的氧化电位,这也是高压 SMBs 实现高能量密度的严峻挑战。尽管在构建合适的SEI方面报道了大量研究性工作,但迄今为止,电解液溶剂化结构中阳离子-溶剂配位和阳离子-阴离子配位对SEI结构的影响很少被研究和报道。事实上,SEI的组成和分布对溶剂化结构中阳离子-溶剂和阳离子-阴离子之间的竞争配位高度敏感。
研究简介
近日,中国科学院金属研究所的杨慧聪、李峰团队基于焓变影响配位的本质,通过调控钠离子竞争配位的策略设计了一种具有稳定电极-电解液界面、高氧化稳定性、快速钠离子输运动力学等综合性能优异的新型电解液体系。通过对溶剂氟取代后,钠离子-溶剂的结合较弱,形成钠离子-溶剂配位结构比形成钠离子-阴离子配位结构导致的焓释放更少。此外,这种低焓的配位结构可以提高竞争配位平衡对盐浓度的敏感性,即使在电解液中盐浓度相对较低的情况下,原始的竞争配位平衡也会发生变化,使更多的阴离子在溶剂化鞘中进行配位。因此,该电解液中Na||Cu电池具有98.0%的高库伦效率,Na||NVPF电池具有优异的循环稳定性,在0.5/1C下500次循环后平均CE 99.4/99.6%,容量保持率为94.3/94.1%。该文章以 “Competitive Coordination of Sodium Ions for High-Voltage Sodium Metal Batteries with Fast Reaction Speed”为题发表在Journal of the American Chemical Society上。
图文导读
1. 溶剂设计对钠离子的竞争性协调调控
如图1a所示,与DEC和EC相比,ETC和FEC中氧原子周围的负电荷较少,导致Na+与溶剂之间的配位较弱(图1b)。与纯溶剂相比,Na+-溶剂的HOMO能级较低,且Na+- PF6-负能级最大(图1c),说明ETC和FEC溶剂分子的加入以及同时增强的Na+- PF6-配位可以获得更高的电解液氧化稳定性 (图1d)。拉曼光谱表明随着氟化水平的增加,1 M NaPF6-DEC/FEC(DF电解液)和1 M NaPF6-ETC/FEC(EF电解液)的PF6-峰分别蓝移至739.7 cm-1和741.5 cm-1,表明阴离子参与溶剂化结构增加。还考察了不同氟化水平的结构对盐浓度的敏感性。当钠盐浓度从1 M增加到1.3 M时(图1e), DEC/EC电解液(DE电解液)中没有观察到PF6-阴离子的蓝移或红移,保持了原有的溶剂化结构。而1.3 M NaPF6-ETC/FEC中PF6-峰呈现蓝移。由于溶剂化结构变化对盐浓度的敏感性,EF电解液中盐浓度的轻微增加可以进一步促进电极上阴离子衍生的界面化学反应。傅里叶变换红外光谱(FTIR)中,Na+-溶剂与游离溶剂的比例随着−F基团的存在而降低:Na+-ETC(12.3%)<Na+-DEC(22.4%),Na+-FEC(72.9%)<Na+-EC(76.8%)。因此两种氟化溶剂的EF电解液表现出优化的溶剂化结构,总体Na+-溶剂配位降低, Na+-阴离子配位增加,图1f中 19F核磁共振(NMR)光谱的化学位移变化进一步验证了这一点。
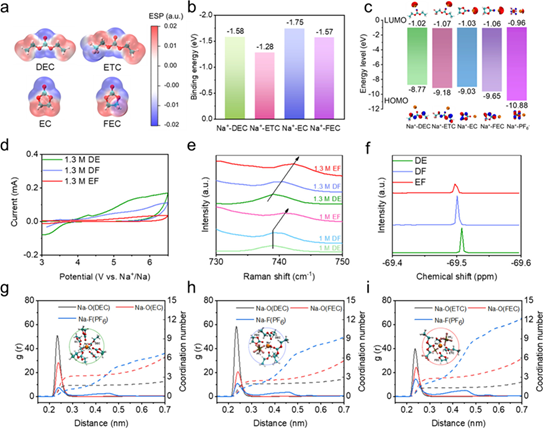
图1:(a)不同溶剂分子的静电势(ESP)。(b) Na+与不同溶剂的结合能。(c) Na+-溶剂和Na+-阴离子的HOMO和LUMO能级。(d) 1.3 M电解液的氧化稳定性的CV测试。(e)不同电解液体系中Na+-PF6-的配位。(f) DE、DF和EF电解液的19F NMR谱。(g) DE, (h) DF和(i) EF电解液的径向分布函数和代表性溶剂化结构(插图)。
为了深入了解溶剂氟化对钠离子竞争配位的影响,进行了经典分子动力学(MD)模拟。当EC被FEC取代时,Na+-FEC表现出比Na+-EC更长的相互作用距离和更低的总Na+-溶剂配位数(图1h)。当DEC进一步被ETC取代时, Na+-溶剂的总配位数进一步降低,Na-F(PF6–)和Na-O(FEC)的配位数增加(图1i)。MD模拟结果与拉曼和FTIR光谱结果一致,证实了通过溶剂设计可以调节钠离子的竞争配位,从而实现富含阴离子的溶剂化结构。
2. 钠金属负极的界面分析
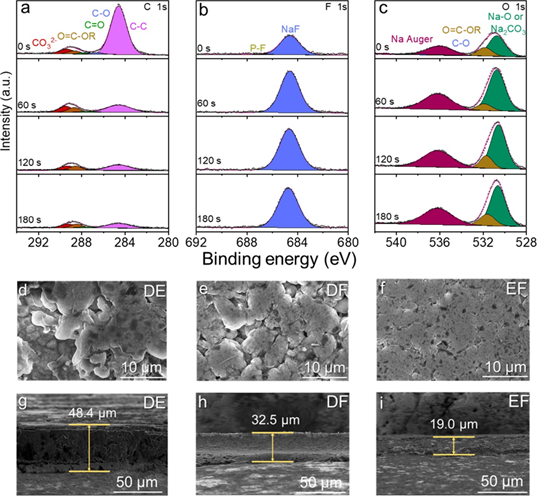
图2.EF电解液在0.5 mA cm–2下循环20圈后的SEI的(a)C 1s、(b)F 1s和(c)O 1s XPS 光谱。在0.5 mA cm –2下(d) DE、(e) DF 和 (f) EF 电解液中沉积钠形态的 SEM 图像。(g-i)在0.5 mA cm–2 下用不同电解液循环后SEI的横截面SEM图像。
采用X射线光电子能谱(XPS)分析不同电解液中SEI的组分,进一步揭示钠离子竞争配位变化与SEI的密切关系。EF电解液中C的原子含量降至最低(7%),F的原子含量增加到12%(图2a)。同时,F 1s谱没有检测到相关的副产物CFx 和P-F物种(图2b),这是由于通过增强Na+-PF6–和Na+-FEC配位,导致FEC和PF6–更彻底地分解。此外,ROCO2Na在SEI内层可能转化为Na2O或Na2CO3,从而形成更多无机组分Na2O(图2c)。由于DE和DF电解液中 Na+-溶剂配位较强,更多的溶剂在Na/电解液界面被还原和不完全分解,从而产生有机主导且更厚的SEI。相反,结合EF电解液中Na+-溶剂配位的降低和Na+-阴离子配位的增加,降低了溶剂分解,获得了富含无机物且坚固的SEI,有利于抑制钠枝晶的形成,加速Na+在SEI中的迁移,从而实现钠的均匀沉积。通过扫描电子显微镜(SEM)观察铜箔上钠金属电沉积的形貌。在EF电解液中观察到沉积钠金属更加平坦均匀,没有枝晶或死钠(图2f),在20次循环后,具有更低的厚度(19.0μm,图2i)。这些事实证实,调节钠离子的竞争配位以获得修饰的溶剂化结构可以形成高度稳定的SEI,以抑制枝晶和副反应。
3. 钠金属负极的沉积/剥离可逆性
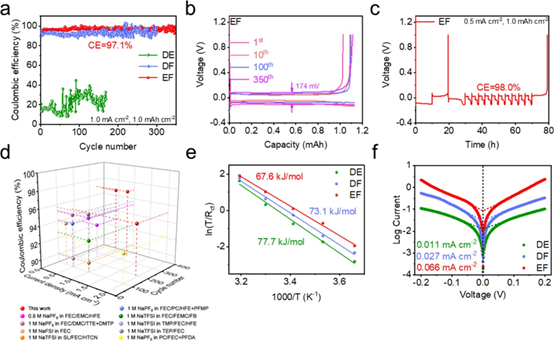
图3.(a)Na||Cu电池在1.0 mA cm–2 –1.0 mAh cm–2下的CE和(b)EF电解液的电压曲线。(c)采用改进方法测试EF电解液在 0.5 mA cm–2 –1.0 mAh cm–2 的CE。(d)与已报道的酯类电解液中Na||Cu电池的CE对比。(e)不同电解液的去溶剂化活化能和(f)交换电流密度。
组装Na||Cu电池以评估钠金属负极的可逆性和电镀/剥离的稳定性(图3a) DE 电解液在 100 次循环中显示出 21.7% 的低平均 CE。在FEC存在的情况下,DF电解液的平均CE明显更高,约为92.7%,表明FEC在循环过程中对SEI有积极影响。此外,EF电解液的平均CE达到97.1%,极化电压较低(第350次循环为174 mV;图3b)。采用改进后的CE测试方法更准确地评估钠电镀/剥离的可逆性,从而获得了98.0%的高CE(图3c和图3d)。EF电解液的对称电池在0.5 mA cm–2 –1.0 mAh cm–2 和1.0 mA cm–2 –1.0 mAh cm–2下可以稳定循环超过1400小时和1000小时。此外,EF电解液对Na+的去溶剂化具有最低的活化能,证实了快速去溶剂动力学(图3e)。此外,随着电解液溶剂氟化水平的增加,可以观察到交换电流密度从0.011 mA cm–2 (DE)逐渐增加到0.027 mA cm–2(DF)和0.066 mA cm–2(EF),如图3f所示。结果表明我们通过溶剂设计来调节钠离子竞争配位的策略成功地优化了溶剂化结构,从而产生了稳固的SEI、优异的循环稳定性和快速的电化学动力学。
4. Na||NVPF电池的电化学性能
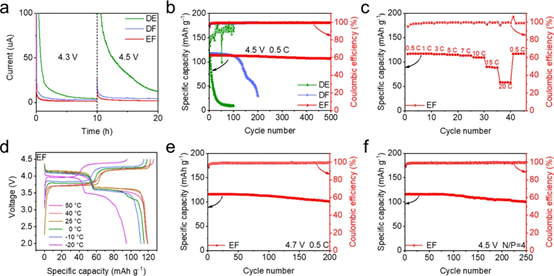
图4.(a)Na||NVPF电池在4.3和4.5V恒定电压下的CA曲线。(b)4.5V、0.5C下Na||NVPF电池的循环测试。(c)4.5V下EF电解液的倍率性能测试。(d)−20至50°C,4.5V、0.5C下EF电解液的充放电曲线。(e)4.7V、0.5C下EF电解液的Na||NVPF电池循环稳定性。(f)4.5V、0.5C下EF电解液Na||NVPF全电池测试(N/P=4:1)。
鉴于钠金属负极在EF电解液中的优异性能,我们组装了Na||NVPF电池,用于评估所设计的电解液在高电压(≥4.3 V)的全电池中的实用性。首先如图4a所示,当电压保持在4.3 V时,与DE(4.4 μA)和DF(3.0 μA)电解液相比,EF电解液表现出最小的漏电流(1.3 μA)和最短达到稳态时间,并且在4.5 V时观察到相同的趋势。这些结果证实了EF电解液在NVPF阴极上具有最高的氧化稳定性和最小的副反应,与DFT计算和CV测试的结果一致(图1c,d)。在 4.3 V 和 0.5 C 下采用EF电解液的Na||NVPF电池具有低电压极化和出色的循环稳定性,在500次循环中容量保持率为95.7%。4.5V下,采用EF电解液的Na||NVPF电池平均CE超过99.4/99.6%,在0.5/1C下500次循环后容量保持率为94.3/94.1%(图4b)。此外,由于快速的钠离子传输动力学, EF电解液表现出优异的倍率性能和宽温性能(图4c和图4d)。即使在 4.7 V 的超高截止电压下, EF电解液的Na||NVPF电池也可以实现超过200圈的稳定循环(图4e)。此外,组装了N/P比为4:1的 Na||NVPF全电池,在 250 次循环后仍具有初始容量的 87.1%(图4f)。
5. 钠离子竞争配位的本质

图5.(a、b)Na+-溶剂和Na+-阴离子之间竞争配位平衡的示意图。Na+-溶剂和Na+-阴离子结合能的相对变化使溶剂化结构的竞争配位平衡从Na+-溶剂向增加的Na+-阴离子转变,这是由焓变决定的。
实际上,SMBs电解液中不同溶剂化结构的比例是由阳离子-溶剂和阳离子-阴离子之间的竞争配位决定的。而这种竞争配位平衡本质上是由形成不同溶剂化结构时的焓变决定的。Na+-溶剂或Na+-阴离子配位结构最终形成取决于哪种配位结构可以实现最小的吉布斯自由能。电解液的吉布斯自由能随Na+-溶剂和Na+-阴离子的含量而变化,即Na+-溶剂或Na+-阴离子配位结构形成时释放的焓变,而焓变由结合能决定。如图6a所示,当使用EC和DEC溶剂时,Na+-溶剂的高结合能通过形成Na+-溶剂配位结构而比形成Na+-阴离子配位结构引起更多的焓释放。因此,DE电解液中的溶剂化结构以Na+-溶剂配位为主,以实现最小的吉布斯自由能。然而,在溶剂的氟取代下,Na+-溶剂较弱的结合通过形成Na+-溶剂配位结构比形成Na+-阴离子配位结构导致的焓释放更少(图5b)。此外,这种释放焓较少的Na+-溶剂配位结构可以提高竞争配位平衡对盐浓度的敏感性。即使EF电解液在相对较低的盐浓度下,原始的竞争配位平衡也会发生变化,使更多的阴离子在溶剂化鞘中配位。这种调控阳离子竞争配位平衡的策略不仅适用于钠金属电池的电解液设计,而且可以扩展到其他具有相同热力学本质的电池体系中的电解液溶剂化结构设计。
总结和展望
综上所述,本文基于焓变影响平衡的性质,设计了一种新电解液体系,实现了具有快速电化学动力学和优异循环稳定性的高压钠金属电池。通过调节钠离子的竞争配位,获得了具有显著阴离子配位的溶剂化结构,从而形成富含无机物的电极-电解液界面、高氧化稳定性和快速去溶剂化动力学。Na||NVPF 电池在 4.5 V 的高截止电压下具有出色的倍率能力、宽工作温度范围和长期循环稳定性。所提出的通过调节阳离子的竞争配位来改变溶剂化结构的策略,为实现稳定的高能量密度钠金属电池提供了新的方向,并且可以通过控制溶剂化结构的焓变进一步扩展到其他电池体系。
审核编辑:刘清
-
轮毂电机驱动电动汽车垂向动力学控制研究综述2025-03-07 561
-
电力拖动系统的动力学课件2008-11-19 4501
-
[下载]想了解多体动力学软件吗?有教程分享及免费试用下载2009-03-24 4379
-
基于多体系统动力学的空气悬架大客车平顺性试验仿真研究2009-12-02 4143
-
汽车系统动力学长篇大论2017-11-01 5155
-
Aigtek高压放大器在压电双晶片动力学研究中的应用2018-11-07 3521
-
飞行器动力学参数在线辨识EKF算法实验流程2021-08-27 1488
-
分布式驱动电动汽车的动力学控制有哪几种类型?常见问题是什么?2021-08-30 1424
-
热分析动力学2009-12-01 737
-
飞盘空气动力学2016-12-25 1702
-
电动力学或经典电动力学统称为什么?2020-06-10 5120
-
电池锂动力学过程中时域分析的理论基础2022-07-10 5526
-
具有高硫载量和高效转化动力学开发RT-Na/S电池应用2022-11-21 1889
-
基于车辆动力学模型的横向控制2023-11-15 2288
-
刚性机械臂的动力学建模2023-11-17 2202
全部0条评论

快来发表一下你的评论吧 !
