

高稳定性富锂锰基正极材料
描述
研究背景
富锂锰基层状氧化物(Li1+x[NiMnCo]1-xO2,LMR-NMC)具有氧阴离子氧化还原的额外容量和出色的价格竞争力,因而是下一代锂离子电池(LIBs)的潜在正极材料。然而,因为在长期循环过程中电压持续衰减,导致能量损失,其商业化之路面临挑战。电压衰减是由于层状相向尖晶石或岩盐相变源于晶格中氧空位的形成和积累,如氧释放,以及金属-氧配位导致的过渡金属(TM)阳离子的无序化等,这些都与氧阴离子氧化还原在热力学上相关。所以,精确理解LMR-NMC的结构演化机制对于稳定长期循环的正极电位至关重要。在纳米尺度上,LMR-NMC的相变往往导致尖晶石-岩盐混合相的形成,使相变途径变得模糊。此外,还没有理论解释局部阳离子迁移、氧演化和远程原子重排序之间的关系,这使得相变机制更难以捉摸。为了解决这些问题,作者将重点放在了LMR-NMC的相变行为的报道差异性,在大多数情况下,观察结果显示尖晶石和岩盐纳米畴在表面共存,形成的尖晶石相在低电位区激活Mn3+/Mn4+氧化还原(《 3.5 V),岩盐相由于缺乏Li扩散路径而产生高过电位,共同导致放电电压下降。此外,De Volder小组最近基于电子显微镜和核磁共振波谱的研究,观察到主要的层状到岩盐表面重建,并保留了原始层状结构。在这种情况下,电压衰减仅仅是过电位增加的结果,平衡电位几乎没有变化。另一方面,Chueh利用纳米级结构和化学探针,提出了层状相中氧空位的积累是Mn氧化还原活化的起源。尽管迄今为止关于LMR-NMC相行为的报道存在显著差异,但还没有系统地尝试控制相变途径,所以本工作通过对岩盐层形成的不稳定性分析,明确地分离了热力学驱动的电压衰减和过电位增加的影响,从而增强了对LMR-NMC电压衰减机制的理解,确立了控制界面氧稳定性是控制整体结构相变和电化学演化途径的关键。
成果简介
近期,韩国浦项理工大学Jihyun Hong教授 在Energy Environ. Sci上重磅发文:“Decoupling Capacity Fade and Voltage Decay of Li-rich Mn-rich Cathodes by Tailoring Surface Reconstruction Pathways”的文章。本工作通过电解质工程揭示了富锂锰基层状氧化物(LMR-NMCs)颗粒表面的相变路径与氧稳定性之间的强烈相关性。作者通过调整表面重建路径,可以控制整体的相和电化学演化机制,从电解质中去除极性碳酸乙烯可显著抑制正极-电解质界面的不可逆氧损失,优先促进原位层状到尖晶石的相变,同时避免典型岩盐相的形成。原位形成的尖晶石稳定表面通过三维离子通道增强了电荷转移动力学,在700次循环中保持了Ni、Mn和O的可逆氧化还原能力。表面尖晶石相导致的深度剥蚀和锂化加速了体层向尖晶石相的转变,引起了热力学电压衰减,但没有容量损失。相反,传统电解质会导致层状到岩盐表面的重构,阻碍电荷转移反应,从而导致容量和电压同时衰减。这项工作解耦了LMR-NMCs中电压衰减的热力学和动力学方面,建立了表面重建、体相变和利用正离子和阴离子氧化还原对的大容量正极电化学之间的相关性,强调了电化学界面稳定对未来锂离子电池中富锰正极化学的重要意义。
图文导读
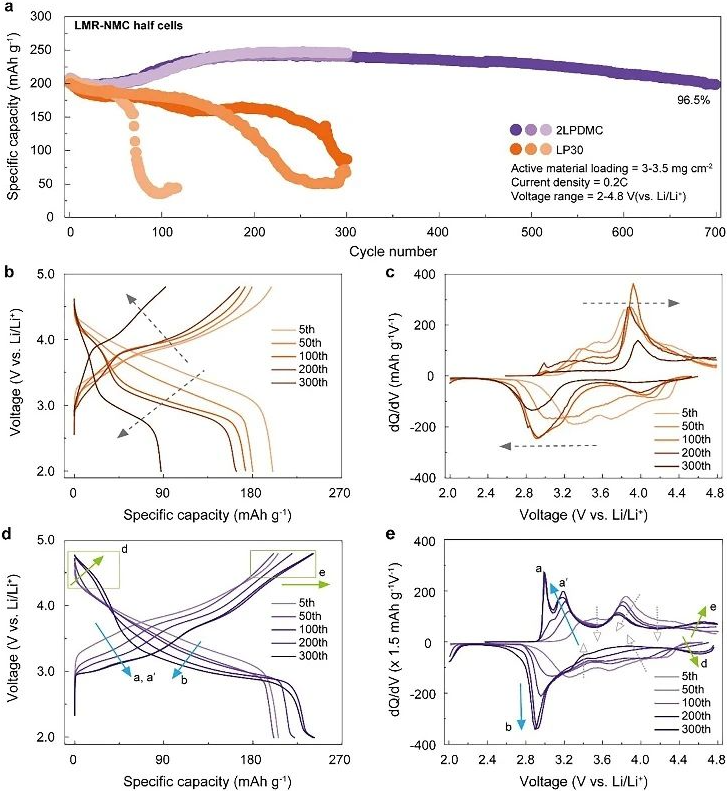
图1. 不同电解质修饰策略调控LMR-NMC的电化学性能
电解液改性解耦过电位和热力学电压衰减分析。为了评估电解质-正极相互作用对电化学的影响,作者合成了初级颗粒直径为100-400 nm的LMR-NMC(化学式为Li1.17Ni0.21Mn0.55Co0.07O2,结晶度高。随后,分别使用1M LiPF6在碳酸乙烯/碳酸二甲酯(EC/DMC, 1:1 v/v,以下为LP30)和2M LiPF6在DMC(以下为2LPDMC)电解质组成的半电池测试了LMR-NMC的长期电化学性能(图1)。结果发现,去除EC对于实现长期稳定性至关重要,增加盐浓度可以增强无EC的DMC基电解质的离子电导率。在LP30中,LMR-NMC在0.2 C下循环250次后仅保持70%的容量,而在2LPDMC中表现出出色的容量保持能力,在第700个周期时提供了96.5%的容量。更有趣得是,容量在第30次和第200次之间增加,表明在循环过程中额外的氧化还原对逐渐激活。为了解电解质修饰引起的电化学可逆性的差异,作者分析了电压分布的演变。结果显示,LP30中长期循环会显著降低放电电压,而充电电压几乎没有变化,表明极化增加。差分容量与电压(dQ/dV)曲线显示,低于3.8 V的充电容量和高于3.3 V的放电容量明显衰减,导致100次循环后的充放电活动不对称。经过300次循环后,由于严重过电位,LMR-NMC失去了大部分的电化学活性。相反,在2LPDMC中,充电和放电曲线以类似的方式向低电位方向发展,在dQ/dV曲线中显示对称的氧化还原峰,经过长期循环后,可逆氧化还原峰出现在3 V附近,表示Mn3+/Mn4+氧化还原偶联的活化。同时,在4.7 V附近出现了额外氧化还原峰,表明形成了尖晶石状相,具有高电压Ni2+/Ni4+氧化还原活性。与此同时,由于原始层状相的比例减少,在3.2~4.4 V范围内的容量减小。结果表明,热力学势的降低是2LPDMC中电压衰减的主要原因,而过电位的影响最小。2LPDMC第300次循环的充放电平均电位差为0.49 V,相当于LP30的47%,证实了过电位的增加有所减轻。即使在高负载下(~8 mg cm-2),电解质修饰的界面稳定仍可有效地延长LMR-NMC的循环寿命。
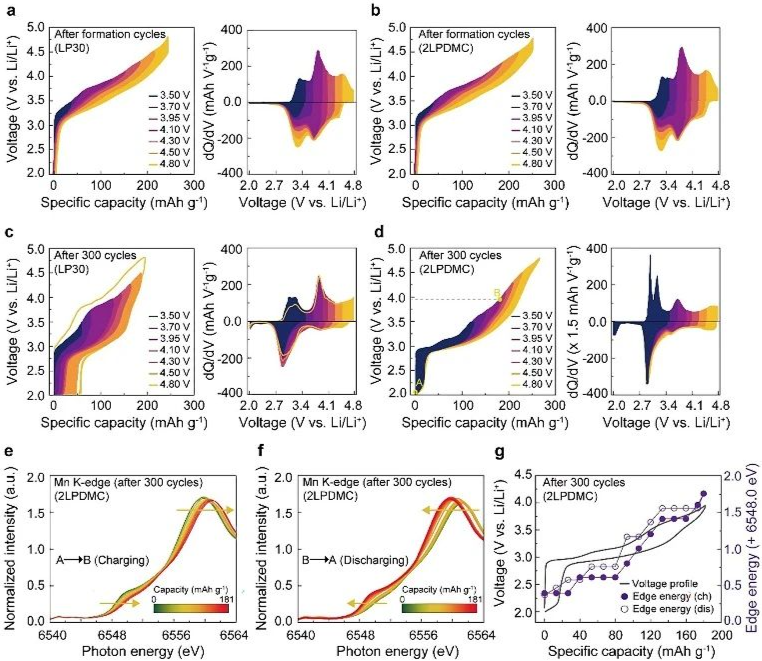
图2. 不同电解质下300次循环后电压迟滞
最小过电位条件下评估长循环后的电压迟滞。改性电解质使过电位最小化可以为在近理想条件下研究LMR-NMC的热力学结构和化学演化机制提供机会,该条件可在较长时间内实现高效、深度锂化和锂化。为了分析两种电解质中氧化还原对及其电位滞后的变化,作者获得了长期循环前后LMR-NMC在电位窗口逐步打开期间的电压分布,除了2LPDMC的容量略高外,LP30和2LPDMC的电压分布差异可以忽略不计。当充电截止电位超过4 V时,在相同电压范围内,负离子氧化还原引起的放电电压曲线表现出明显的滞后性(橙黄色阴影部分),这种迟滞导致了不对称的dQ/dV曲线。因此,在相同锂成分下,放电电压或充电状态(SOC)随着充电截止电压的增加而降低。同样,随着放电截止电压的降低,相同荷电状态下的充电电压也会升高,这是由于在低压状态下氧化氧阴离子的减少,以及阳离子从Li层迁移到TM层所致。
相反,两种电解质的长循环导致了电化学演化途径的差异。经过100次循环后,在2LPDMC中循环的LMR-NMC正极在3.0 V附近的充电和放电均表现出明显更高的氧化还原活性,与LP30相比具有更小电压滞后。3.5 V下的低迟滞表明可逆阳离子氧化还原的贡献更大,这是由于Mn3+/Mn4+氧化还原偶联的激活。经过300次循环后,LP30中的LMR-NMC发生了严重的充放电极化,导致显著的电化学不可逆性。值得注意的是,一旦充电超过4.5 V,利用阴离子氧化还原,过电位急剧增加,限制了下一个周期的容量,如在4.5-2.0 V周期后测量的黄色曲线。这一结果将商业电解质中容量保持差归因于由过电位引起的动力学驱动的电压滞后。另一方面,在2LPDMC中,LMR-NMC在300次循环后仍保持了良好的电化学可逆性,在dQ/dV曲线上呈现出尖锐而对称的氧化还原峰。循环正极分别在3.0 V和3.2 V处显示出两个不同的电压平台,充电和放电之间的电压间隙很小,表明阳离子主导氧化还原贡献。在2~3.9 V之间的原位X射线吸收近边结构(XANES)光谱明确证明Mn氧化还原是可逆的。dQ/dV曲线显示,在充电至4.8 V后,氧化还原峰出现在4.5 V以上,这可能是由于高压尖晶石氧化物如LiNi0.5Mn1.5O4(LNMO)的形成,其中Ni2+/Ni4+具有活性。与之前的氧阴离子氧化还原反应相比,新的氧化还原反应表现出低电压滞后性,表明了阳离子主导的氧化还原机制,结构无序性可以忽略不计。先前使用商业电解质的大多数研究可能难以观察到尖晶石Ni在充电结束时的氧化还原活化,因为过电位显著增加。恒流间歇滴定技术(GITT)进一步证实了LP30的缓慢动力学限制了循环过程中LMR-NMC的分解深度,从而抑制了LMR-NMC的结构演变,而2LPDMC的反应动力学增强则加速了结构演变,导致了热力学势的衰减。
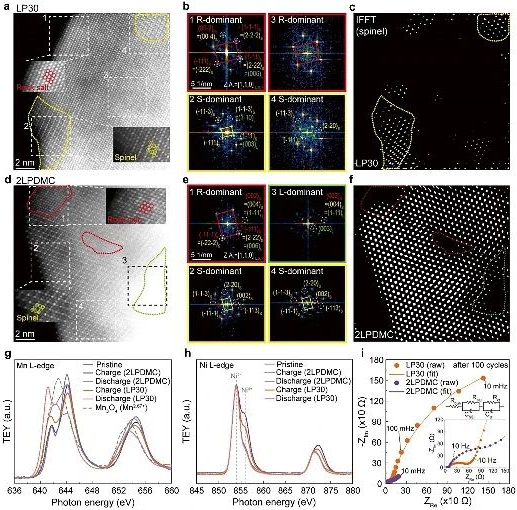
图3. 减少表面氧损失的LMR-NMC定制表面重建途径表征
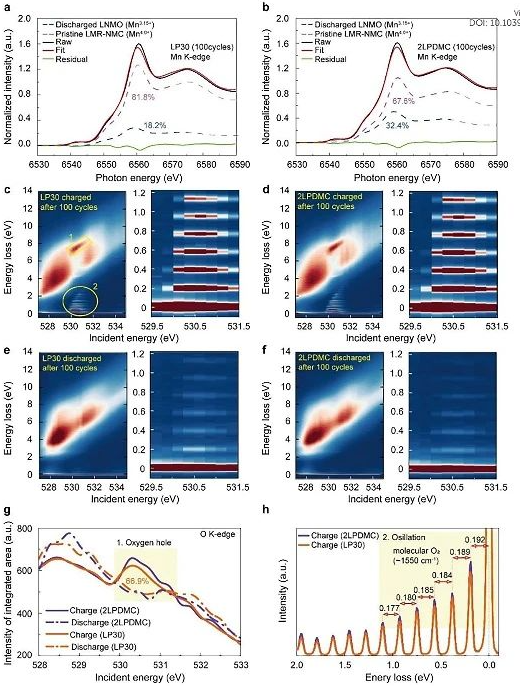
图4. 100次循环后LMR-NMC电极的同步辐射
LMR-NMC的定制氧化还原途径分析。随后,作者分析了循环LMR-NMC的平均TM氧化态,以评估不同结构转变途径下氧化还原演化的差异。从Mn K-edge XANES透射光谱可以看出,经过短期循环(20次),LMR-NMC正极在两种电解质中表现出相似程度的Mn还原,即Mn氧化还原活化。100次循环后,LP30的Mn3+/Mn4+活化进展不大。而在2LPDMC中,经过300次循环后,Mn的还原程度更高,尖晶石区向整体的扩展也更明显,线性组合拟合结果显示,经过100次循环后,2LPDMC和LP30的放电LMR-NMC中Mn4+的残留比例分别为67.6%和81.8%。在具有尖晶石优势表面的LMR-NMC中,Mn的氧化还原活性较高,证实了从表面到颗粒中心的层状到尖晶石相变的穿透性,如STEM-HAADF图像所示。相反,充电正极在100次循环后的Mn-K边缘光谱显示出可以忽略不计的差异,这表明岩盐相分数由于其在CEI附近的空间约束形成而最小。同时,循环后两种电解质的Ni氧化态仅相差1.5%,可以忽略不计。因此,LP30的容量衰减主要是由于动力学迟缓的阴离子氧化还原活性的丧失,这伴随着晶格内过渡金属的迁移。然而,在LP30中长期循环后,即使在高过电位的LMR-NMC中,几乎完全获得Ni氧化还原能力也是可能的。为了评估循环后阴离子的氧化还原活性,在两种电解质中循环100次后,收集了正极O K边缘的高分辨率共振非弹性X射线散射光谱。在放电的正极中,局部的空穴随着振动信号消失,即使在100次循环后也证明了可逆的氧化还原活性。根据RIXS图谱的定量分析,LP30中100循环LMR-NMC的局部氧孔综合面积为2LPDMC的66.9%,LP30中的分子O2信号强度略低于2LPDMC。然而,在LP30和2LPDMC中,循环正极之间的分子O2振动强度差异并不像局部氧空穴特征那样明显。这一对比表明,氧化阴离子除了以分子形式存在外,还存在多种形式,如金属-氧多键、过氧类二聚体和具有局域空穴的晶格阴离子。LP30中阴离子氧化还原活性较低意味着严重的极化抑制了阴离子氧化还原,其中缓慢的动力学使阴离子氧化电位升高到超过截止电压。当充电至5.0 V时,可以进行阴离子氧化还原,严重过电位导致的氧化还原活性受到抑制,最终导致电池在长时间运行时容量迅速衰减。
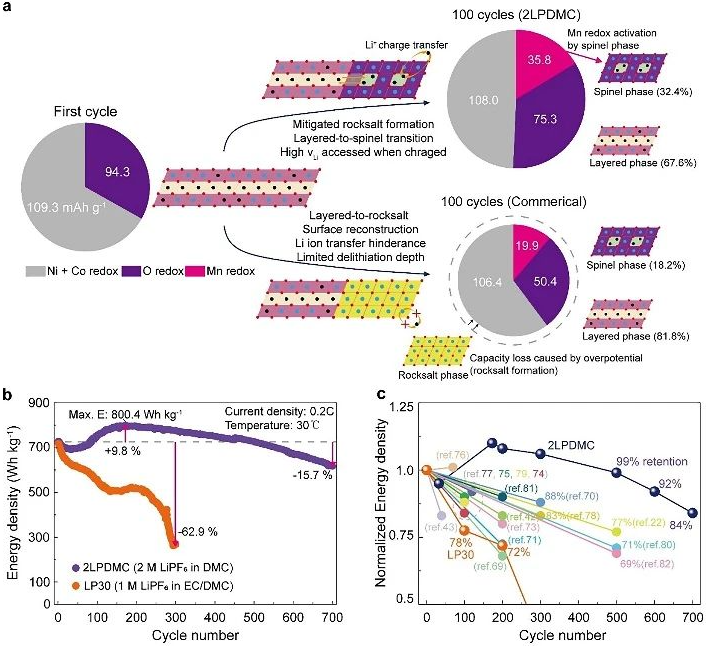
图5. LMR-NMC不同降解机制分析
稳定正极表面氧化阴离子,促进快速Li+转移反应。最后,作者分析了与初始循环相比,LMR-NMC在两种电解质中长期循环时相和氧化还原对的演化途径。当在LP30中循环时,通过严重的氧损失和晶格致密化发生层状到岩盐表面的重建,除了岩盐相的不氧化还原活性外,高阻抗限制了腐蚀深度,从而降低了带电LMR-NMC中的Li空位浓度。较低的Li空位浓度可能对阳离子迁移和层状向尖晶石转变提供的驱动力不足,导致Mn活性降低。因此,LMR-NMC在常规电解质中的电压衰减通常伴随着持续的容量损失,这仅仅反映了过电位的增加,而不是平衡电位的变化。因此,解释电压衰减需要仔细检查充电和放电电压,以解耦热力学和动力学贡献。相反,在2LPDMC中,正极表面经历原子重建,导致尖晶石为主相,在没有EC的电解质中,不稳定的岩盐形成类似于之前观察到的LNMO在没有EC的电解质中保存完好的尖晶石结构。尖晶石相活化的Mn3+/Mn4+氧化还原提供了35.8 mAh g-1的额外阳离子氧化还原容量,补偿了降低的阴离子氧化还原活性。同时,颗粒表面尖晶石相的三维互联锂通道可能促进电化学界面上的电荷转移,增加延长循环的实际容量。结果表明,原位形成尖晶石表面后,LMR-NMC在100次循环后表现出比2LPDMC初始状态更好的倍率能力。尽管热力学电压衰减,电化学活性的增强提高了能量密度,从699.3 Wh kg-1增加到800.4 Wh kg-1,在500、600和700个循环后,保留率分别为99%、92%和84%。相同的正极在常规电解质中循环300次后,能量保留率仅为37.1%。这项研究表明,在电化学界面上稳定尖晶石相和岩盐相是实现锂离子电池中高能量和长循环寿命富锂/锰正极化学的有力战略途径。
总结与展望
综上所示,考虑到金属-氧配位、TM迁移和氧阴离子氧化还原通过配体-金属电荷转移之间的因果关系,逐渐的电压和容量衰减被认为是LMR-NMC利用阴离子氧化还原获得高容量的必然结果。通过稳定层状到岩盐表面重构的层状-顶尖晶石相变路径,作者明确地表明电压衰减不一定会引起阻抗增加和容量损失。通过调节电化学界面的稳定性,区分了结构演化途径,成功地分离了表观电压衰减的热力学和动力学贡献。与传统的降解机制相比,尖晶石稳定的表面重构增强了电化学反应动力学,实现了高能量密度保留和高速率能力,同时在700多个循环中高效地利用阳离子和阴离子氧化还原。了解层状相、尖晶石相和岩盐相的相对稳定性与电解质化学和物理性质之间的关系,是控制表面重构速度和方向的一个重要课题,这将在接下来的研究中进行。这项发现提出了一种有效的策略,可以实现可行的富锂锰基下一代正极材料,这将是朝着具有成本效益和可持续可充电电池迈出的重要一步。
文献链接
Decoupling Capacity Fade and Voltage Decay of Li-rich Mn-rich Cathodes by Tailoring Surface Reconstruction Pathways. Energy Environ. Sci., 2024. (DOI:10.1039/D4EE02329C);https://doi.org/10.1039/D4EE02329C.
-
定量识别掺杂位点:解锁富锂正极材料的高性能与稳定性2024-12-09 2645
-
利用太阳辐射直接修复富锂富锰正极!2024-03-11 4062
-
锂锰电池的正极材料是由什么组成的?锂锰电池正极材料的优点2023-11-10 2020
-
锰酸锂正极材料的特点和优点2022-08-29 13250
-
全面剖析富锂锰基正极材料!2021-05-10 45493
-
四大动力电池正极材料性能对比2021-03-18 3445
-
四大动力电池正极材料性能的对比2020-12-25 3683
-
揭秘锂电池富锂锰基正极材料的研究进展2019-07-21 10627
-
我国研发出富锂锰基动力电池正极材料 1380Wh/kg以上的比能量密度2018-11-23 3283
-
锂离子电池的最新正极材料:掺锰铌酸锂?2016-01-19 6468
-
锂离子蓄电池正极材料尖晶石型锰酸锂的制备2011-03-10 2780
-
锂电池正极材料锂锰氧化物的改性与循环寿命2009-11-03 541
-
钴镍锰(三元)正极材料---钴酸锂的理想替代品2009-10-29 2493
全部0条评论

快来发表一下你的评论吧 !
