

高通量测序生物信息学分析
电子说
描述
高通量测序技术产生的DNA序列数据长度较短,而且数据量非常巨大。分析了高通量测序环境下大数据的挑战和机遇,总结并讨论了数据压缩、宏基因组数据序列拼接、宏基因组数据序列分析方面的算法和工具等研究成果。最后,展望了高通量测序下DNA短读序列数据研究的发展趋势。
高通量测序分析
高通量测序,一次性对几百万到十亿条DNA分子进行并行测序,又称为下一代测序技术,其使得可对一个物种的转录组和基因组进行深入、细致、全貌的分析,所以又被称为深度测序。主要包括:High-throughput Sequencing,Next Generation Sequencing,Deep Sequencing。
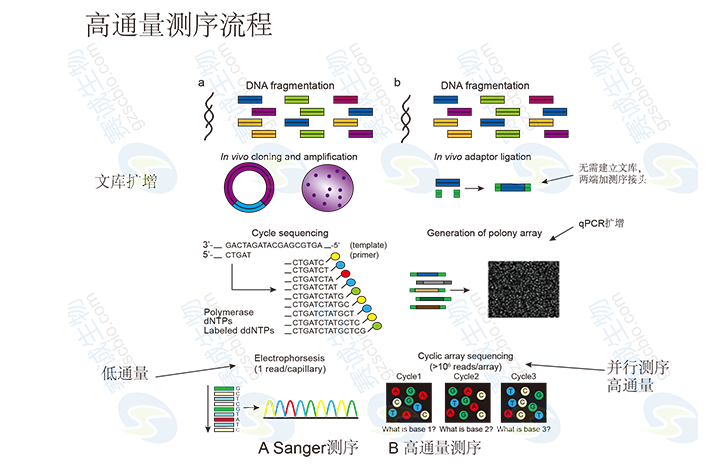
图1 高通量测序流程
高通量测序应用范围广泛:
1 DNA测序:全基因组de novo测序,基因组重测序,宏基因组测序,人类外显子组捕获测序。
2 RNA测序:转录组测序,小RNA测序,电子表达谱测序。
3 表观基因组研究:ChIP-Seq,DNA甲基化测序。
基因组测序
基因组测序是对物种的基因组DNA打断后进行高通量测序,根据是否有已知基因组数据主要分为de novo全基因组测序和基因组重测序。De novo 基因组测序是对未知基因组序列的物种进行基因组从头测序,利用生物信息学分析手段对序列进行拼接、组装,从而获得该物种的基因组图谱。全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
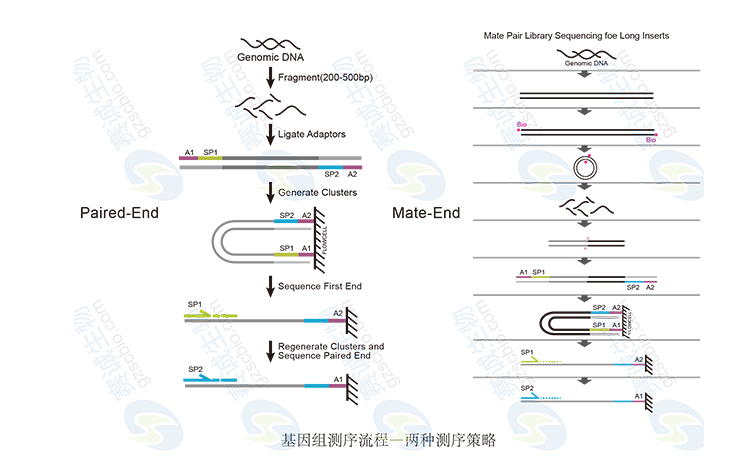
图2 基因组测序策略
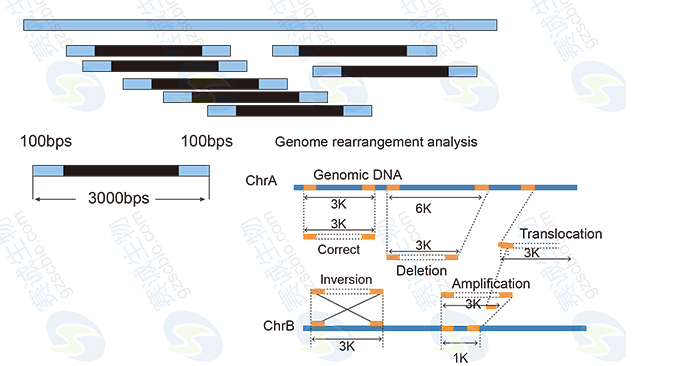
图3 Paired-end原理
Paired-End方法,基因组打断后,选择一定长度(200-500bp)的序列连接两端接头进行两头测序。Mate-end建库较复杂,序列打断后,选取一定长度序列(3-5kb),需先连接生物素,再环化,再打断,生物素富集,连接两端接头进行两端测序。
基因组测序应用生物信息学分析其结果,主要涵盖以下几方面。
1 数据产出处理:图像识别与Base Calling\去除接头序列、检测与去除污染序列等;
2 基因组组装:原始数据统计、测序深度分析、组装结果统计等;
3 基因组注释:Coding Gene注释、RNA分类注释、重复序列注释等;
4 基因功能注释:GO功能分类、Interpro功能分类等;
5 比较基因组及分子进化分析:SNP/InDel/CNV检测等。
宏基因组测序
宏基因组测序是对某一特定环境,如肠道、土壤、海水等中的所有微生物进行基因组测序。通过此方法可对该环境中的微生物种类和优势物种进行检测,揭示微生物群落多样性、种群结构、进化关系、功能活性、相互协作关系及与环境之间的关系 。自然环境中很多微生物无法分离培养,而此方法无需对微生物进行分离培养。宏基因组测序方法现在有全基因组的宏基因组测序和16S/18S rRNA宏基因组测序。
1 全基因组的宏基因组测序
通过高通量测序技术,对环境样品的总 DNA 直接进行全基因组的宏基因组测序,能够实现微生物群落的物种分类研究、群落结构、系统进化、功能注释以及物种间的代谢网络研究,挖掘具有应用价值的基因资源,开发新的微生物活性物质。与传统的 Sanger法相比,速度快,性价比高,周期短,单个样品的测序量可以接近饱和。
宏基因组测序信息分析主要包括:拼接组装,物种分类组成分析,基因预测和功能注释,生成Profiling table,主成分分析(PCA),筛选与样品分组显著相关的因子,多样品间比较分析等。
2 16S/18S rRNA宏基因组测序
16S/18S rRNA是微生物群落分析和细菌进化研究以及分类研究最常用的靶分子,采用新一代测序技术,对16S/18S rDNA的可变区进行测序分析,不需进行克隆筛选,能全面的反映微生物群体的物种组成,真实的物种分布及丰度信息。
16S/18S rRNA测序信息分析主要包括:物种分类、物种丰度分析,OTU(Operational Taxonomic Units)分析,多样性分析,系统进化分析,多样品间的比较分析等。
人类外显子组捕获测序
外显子组是指全部外显子区域的集合,该区域包含合成蛋白质所需要的重要信息,涵盖了与个体表型相关的大部分功能性变异。与全基因组重测序相比,外显子组测序只需针对外显子区域的DNA,覆盖度更深、数据准确性更高,更加简便、经济、高效。
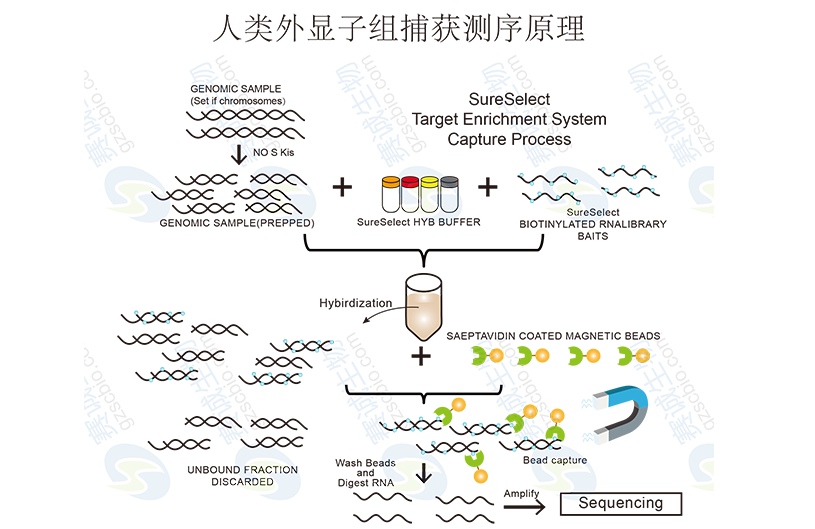
图4 人类外显子组捕获测序原理
外显子捕获是指用外显子芯片杂交,把基因组外显子序列进行捕获,然后对所捕获的序列进行测序。现在常用外显子芯片有Roche NimbleGen Sequence Capture 2.1M Human Exome Array和Agilent SureSelect Target Enrichment System(Human Exome)。
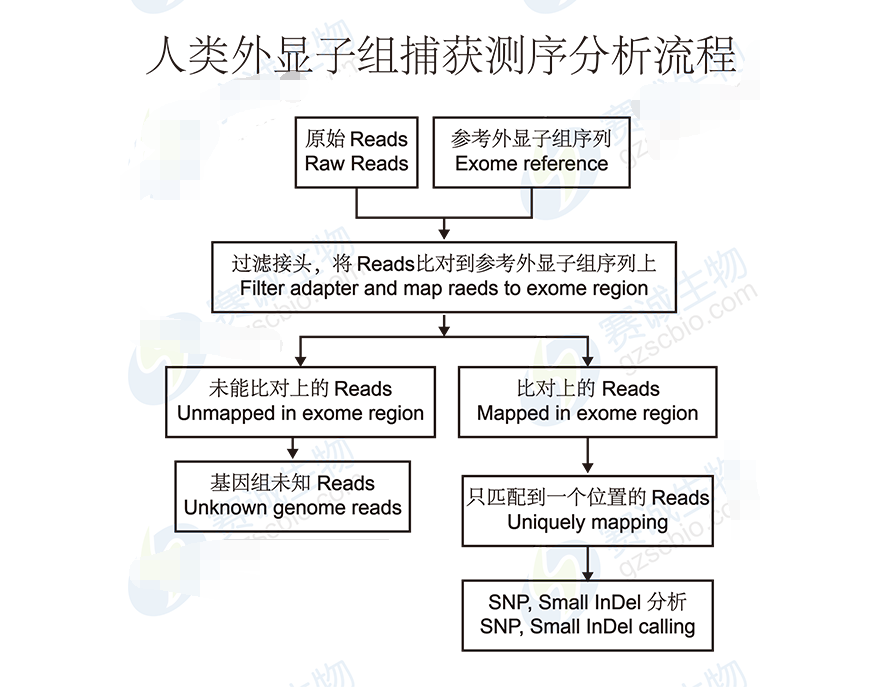
图5 人类外显子组捕获测序分析流程
转录组测序
转录组即特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA(Non-coding RNA)。
第二代测序系统可精确检测单个碱基,并且不受到研究中先验信息的干扰,科研人员能够快速地获得某一物种特定器官或组织在某一状态下几乎所有mRNA转录本序列,从而能够开展:UTRs区域界定、可变剪切研究、低丰度新转录本发现、融合基因鉴定、cSNP(编码序列单核苷酸多态性)研究等。
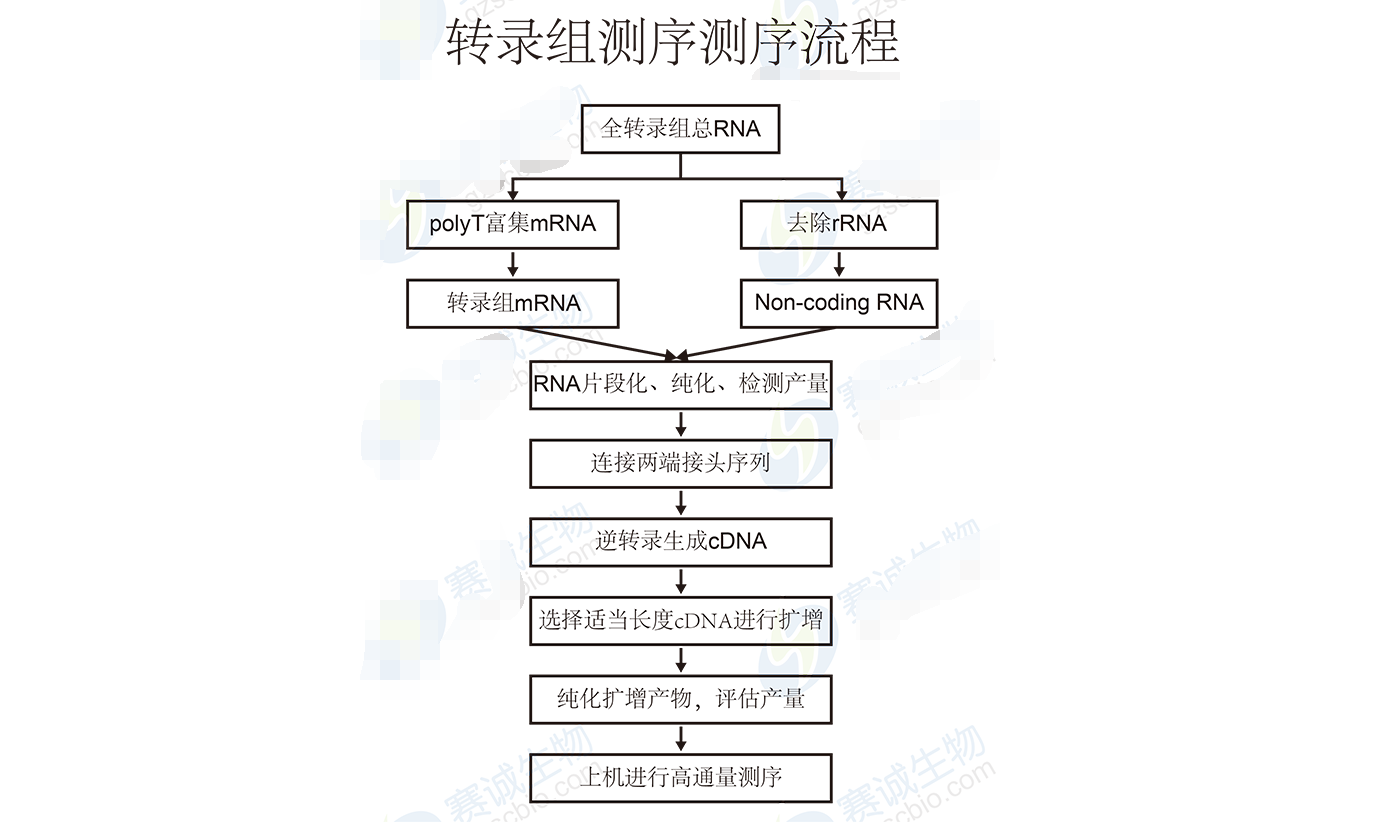
图6 转录组测序流程
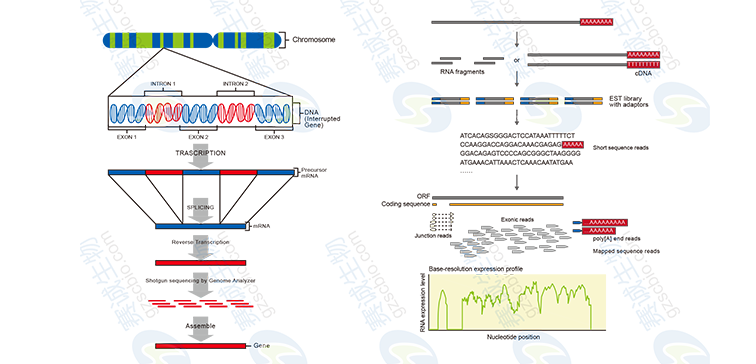
图7 无参考序列及有参考序列转录组测序流程
无参考序列转录组分析内容包括:1 测序数据产量统计,数据成分和质量评估;2 Contig及Scaffold长度分布;3 Unigene的长度分布和功能注释,GO分类,Pathway分析,差异表达分析;4 蛋白功能预测与分类,差异表达基因GO富集和 Pathway富集分析。
有参考序列转录组分析内容包括:1 基本数据统计,比对参考序列;2 序列在基因组上在分布;3 测序深度分析、随机性评估和基因差异表达分析;4 新基因预测,基因可变剪接鉴定和基因融合鉴定等。
电子表达谱测序
电子表达谱测序(Digital Gene Expression, DGE)又称为基因表达标签测序(mRNA tag profiling),又称Tag-SAGE。其原理是通过两种酶切作用对基因中一段长度为21nt的序列标签进行测序。由于其测序只针对表达的基因进行测序,产生的数据量相对较小,是研究基因表达谱的经济而快速的研究手段。是对特定处理条件下的全基因组基因表达谱进行分析,已被广泛用于功能基因组学和医学等研究领域。
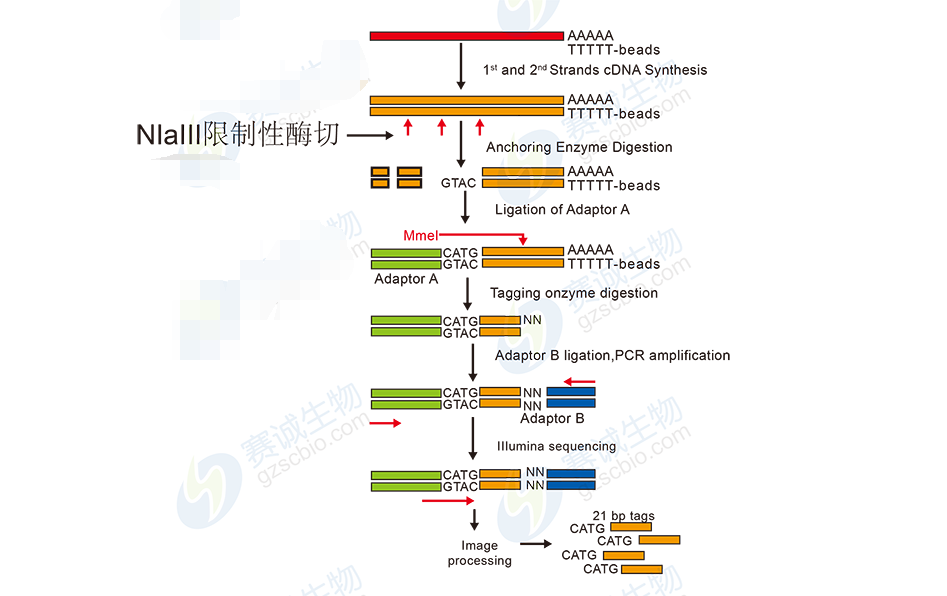
图8 电子表达谱测序流程图
电子表达谱分析内容包括:图像识别与原始碱基数据读取,去污染、去接头,标签序列计数统计,基因组比对与统计,基因序列比对获得所表达的基因列表,基因差异表达分析,聚类与表达类型分析,GO基因富集与分类分析,Pathway富集与分类分析,蛋白相互作用网络分析,反义链转录本与新转录本检测等。
小RNA测序
小 RNA是指长度在21-31nt的内源性非蛋白质编码RNA,广泛存在于高等和低等生物体内,其对mRNA的转录及转录后水平等生命过程起到调节作用。现已知小RNA可归纳成三类:微RNA (miRNA),小干扰RNA(siRNA)和与piwi相互作用的RNA(piRNA)。
miRNA长度为21~24nt,产生于有典型茎环二级结构的原转录本(pri-miRNA),在动植物的目标mRNA的降解与抑制方面发挥重要作用。siRNA,长度在19~25nt,产生于长双链RNA,同样在动植物的目标mRNA的降解与抑制方面发挥重要作用。piRNA,长度26~31nt,由与其相互作用的Piwi蛋白定义,目前研究表明其在配子形成的过程中起作用。
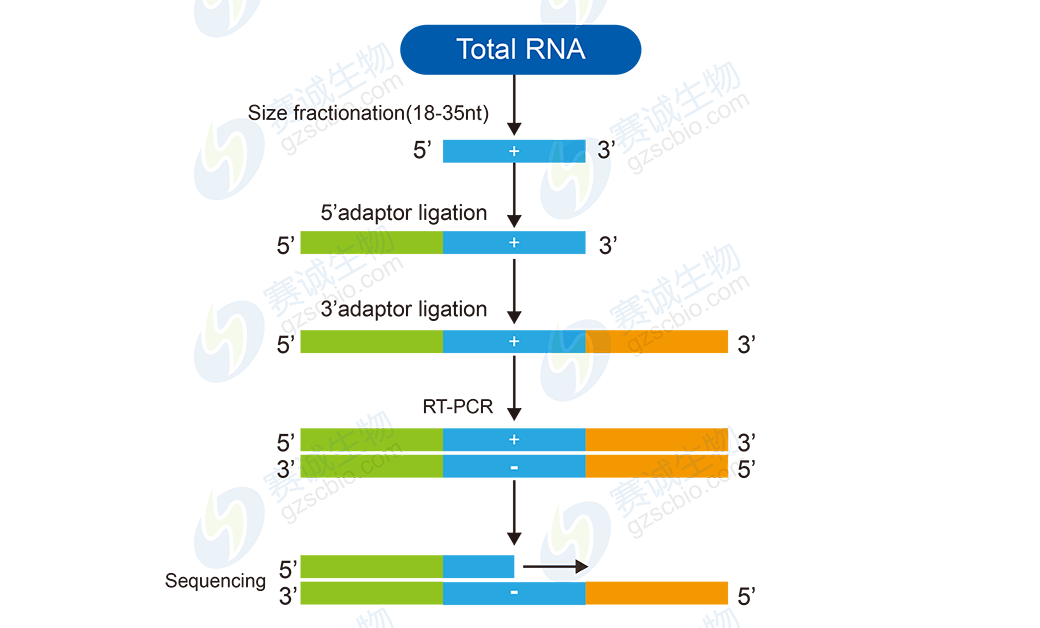
图9 小RNA测序流程图
小RNA测序分析内容包括以下两个主要方面:
1 基本分析:原始数据读取,去接头、去污染序列,长度分布统计,基因组比对等。
2 高级分析:Small RNA的分类注释,miRNA / siRNA / piRNA的鉴定,新miRNA预测,差异表达miRNA聚类分析等。
ChIP-Seq
ChIP-Chromatin Immunoprecipitation染色质免疫共沉淀,是指通过蛋白免疫相互作用,用抗体把和染色质相互作用的蛋白,如组蛋白、转录因子等,沉淀下来,从而所获取与其相结合的DNA序列。ChIP-Seq就是通过高通量测序对ChIP所得到的序列进行测序,从而进行蛋白和DNA相互作用相关研究。
ChIP-Seq分析内容包括:
1 ChIP Sequencing结果与参考基因组序列进行比对。
2 ChIP Sequencing reads 在全基因组的分布:唯一比对reads 在repeats 区域的分布,唯一比对reads 在各基因功能元件上的分布,唯一比对reads 的全基因组覆盖深度。
3 全基因组peak 扫描:peak 扫描,peak 长度分布统计,peak 的全基因组覆盖度,peak 在基因功能元件上的分布特征,
4 Peak相关基因分析筛选与GO功能富集分析。
5 多个样品的差异分析:基于peak 相关基因的差异分析,基于peak 的差异分析。
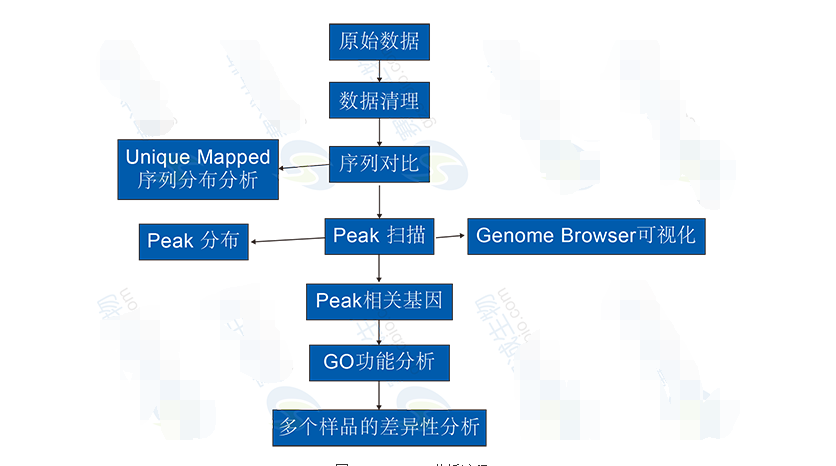
图10 ChIP-Seq分析流程
DNA甲基化测序
DNA甲基化对机体发育和基因表达有很重要的调控作用,和各种癌症的发生和发展也有很大相关性,所以对基因组DNA甲基化进行研究是一直来的热门课题。通过高通量测序来研究DNA甲基化现在主要有两种方法,一种是MeDIP,是通过与DNA甲基化位点相结合的抗体,进行免疫共沉淀,然后对所得DNA序列进行测序。另一种是Bisulfite Sequencing,是通过Bisulfite处理基因组来区分甲基化位点。
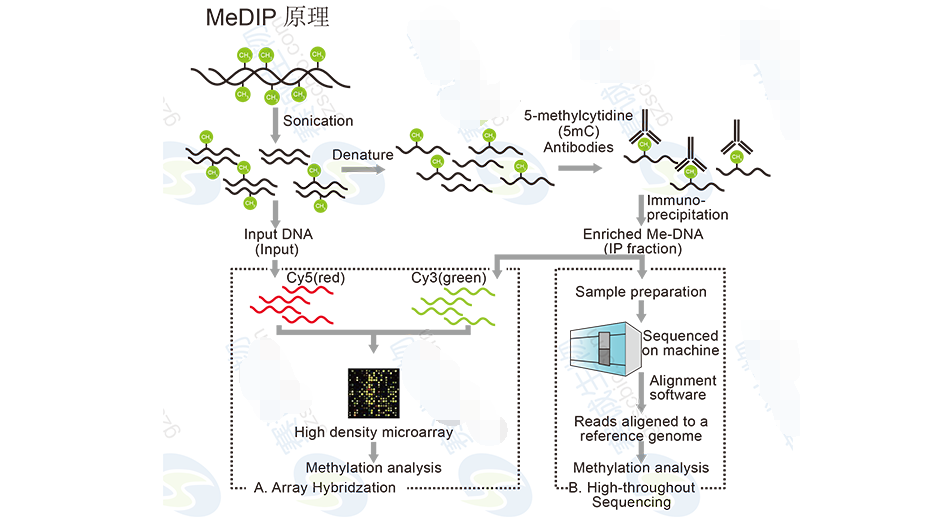
图11 MeDIP 原理
MeDIP-Seq分析内容包括:
1 MeDIP-seq 序列与参考序列的比对。
2 MeDIP-seq 序列数据在全基因组的分布趋势: MeDIP-seq 测序reads 在全基因组上每条染色体上的分布,MeDIP-seq 测序reads 在全基因组上的覆盖深度,MeDIP-Seq 测序reads 在CG、CHG和CHH位点上的覆盖深度,MeDIP-Seq 测序reads 在不同基因功能元件上的分布,MeDIP-Seq 测序reads 在不同OE含量区域中的分布。
3 统计MeDIP-seq 序列富集区域(peak)的信息:Peak 扫描,Peak 长度数量及比例分布统计,单个样品Peak 的OE含量分布统计,寻找Peak 相关基因,统计Peak 在不同基因功能元件上的分布。
4 基于Peak 的多样品间差异分析:分析两个样品间的Peak 相关差异基因,对两个样品间的差异基因进行GO功能富集分析及pathway 功能分析。
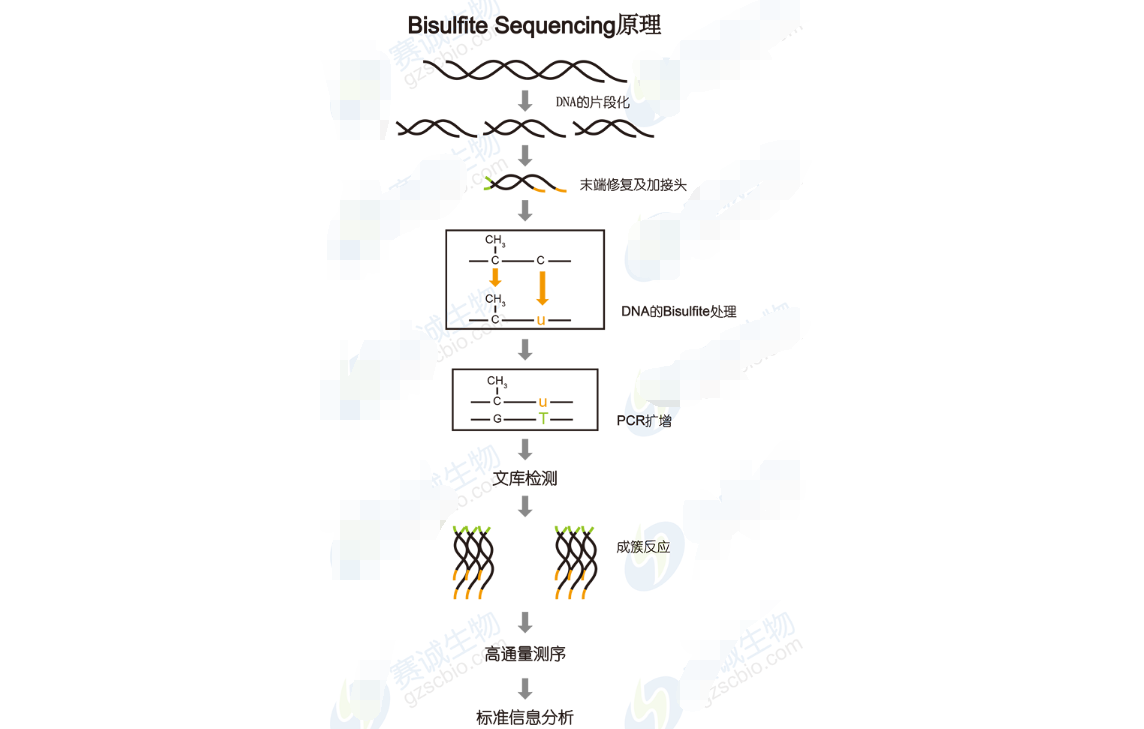
图12 Bisulfite Sequencing原理
Bisulfite Sequencing分析内容包括:
1 Bisulfite-seq序列与参考序列的比对。
2 深度和覆盖度分析:C碱基有效测序深度的累积分布,不同reads 测序深度下的基因组覆盖度。
3 计算C碱基的甲基化水平。
4 全基因组甲基化数据分布趋势分析:甲基化C碱基中CG, CHG 与CHH的分布比例(H=A、C or T),CG、CHG和CHH中的所有C的甲基化水平,各条染色体中CG、CHG和CHH中C的甲基化水平(该项分析目前只用于“人”),统计不同基因区域内CG、CHG和CHH中C的甲基化水平,不同基因元件区域中CG、CHG和CHH中C的甲基化水平,CHG,CHH中甲基化C附近的9bp序列的序列特征分析。
5 全基因组DNA 甲基化图谱:染色体水平的甲基化C碱基的密度分布(该项分析目前只用于“人”),Scaffold的甲基化C碱基密度分布(该项分析针对物种:非人),不同基因组区域的甲基化分布特征,基因组不同转录元件中的DNA甲基化水平。
6 差异甲基化区域(DMR)分析。
- 相关推荐
- 热点推荐
-
超集信息亮相 NCCBB 2026:液冷异构算力,为生物信息研究按下 “加速键”2026-05-28 248
-
高通量生物分析技术之微流控芯片2024-11-14 1622
-
使用北鲲云在AWS上运行基因分析HPC任务2022-11-16 1335
-
用NVIDIA Clara Parabricks v4.0大众化和加速基因组测序分析2022-10-11 3012
-
全基因组测序的优势 精选资料分享2021-07-29 1742
-
高通量测序数据分析:RNA-seq 精选资料分享2021-07-26 1567
-
披荆斩棘,乘风破浪——真迈生物高通量基因测序仪GenoLab发布2020-10-21 3234
-
生物信息学算法导论(脑控技术丛书)2020-02-19 2642
-
什么是高通量单细胞RNA测序技术?2019-04-25 11580
-
Clay Breshears博士讨论基因组测序和生物信息学2018-11-07 3674
-
高通量测序常用名词汇总2018-02-28 4884
全部0条评论

快来发表一下你的评论吧 !
