

锂电池制造 | 破解石墨/硅复合电极的奥秘:纳米多孔结构设计推动高能量锂电池发展
描述
【美能锂电】在电动汽车革命浪潮中,高能量锂离子电池扮演着关键角色。然而,目前主流的石墨负极材料理论容量有限,仅为372 mAh g⁻¹。科学家们将目光投向了硅材料,其理论容量高达3579 mAh g⁻¹,几乎是石墨的十倍。但硅材料在充放电过程中巨大的体积变化导致容量快速衰减,限制了其在商业电池中的应用。
微观世界的力学博弈
Millennial Lithium
最新发表在《自然·纳米技术》的研究通过多模态原位成像技术,深入揭示了石墨/硅复合负极中的电化学-机械过程。研究团队发现,硅颗粒的电化学循环稳定性强烈依赖于颗粒内部纳米级多孔结构的设计。

石墨/μ-Si复合电极在首次和第二次锂化过程中的原位光学显微镜观察
图中展示了电极在40% SOC、60% SOC和100% SOC状态下的微观结构演变、微分位移场和累积应变场。青色箭头指向两个经历体积膨胀和断裂的代表性硅颗粒。研究显示,在ROI 2区域,被石墨颗粒紧密包围的中央硅颗粒直到90% SOC才被激活,这可能是由于致密化导致的局部电解质不足。
纳米孔隙的关键作用
Millennial Lithium
通过X射线纳米CT技术,研究人员发现商业微米硅颗粒具有单向管状孔隙结构,这些孔隙在材料合成过程中通过定向凝固过程形成。
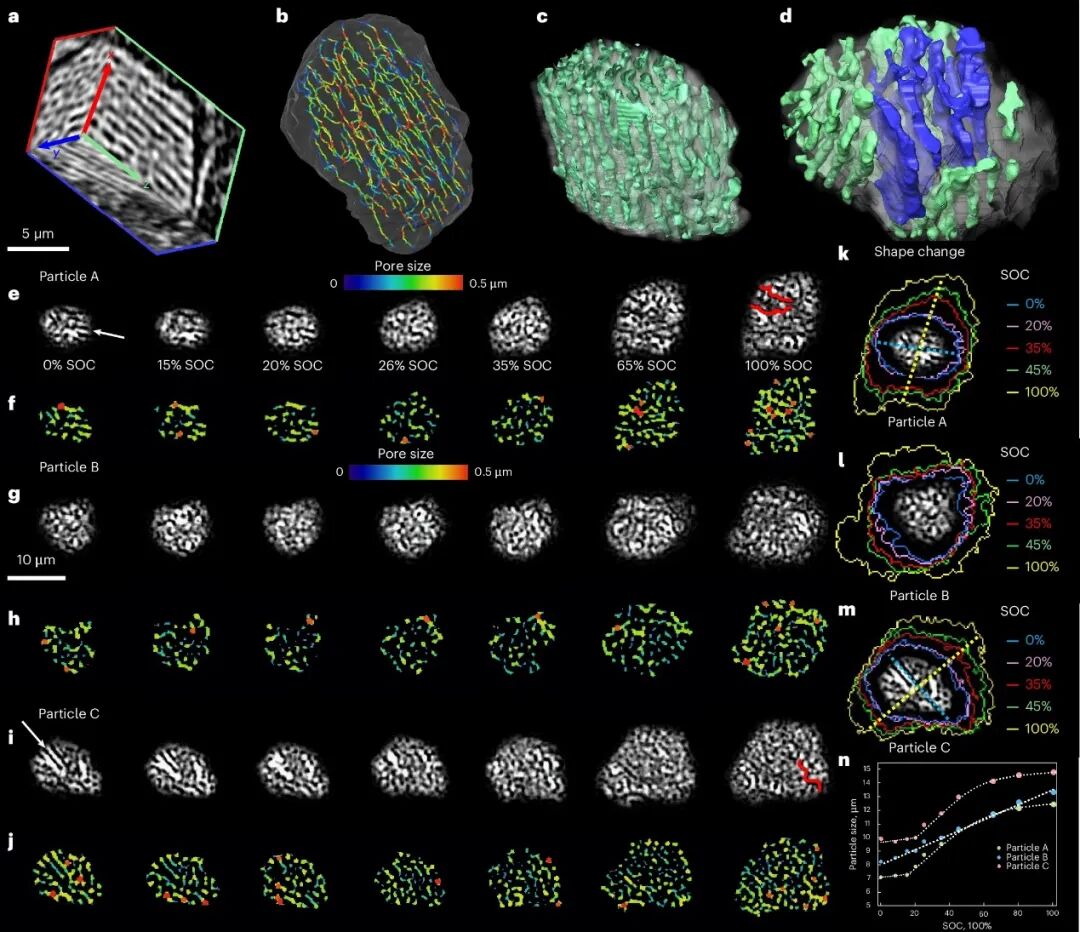
三种不同多孔硅颗粒的微观结构演变
研究跟踪了三种具有不同孔隙结构的硅颗粒:颗粒A具有水平管状孔隙,颗粒B具有点状孔隙,颗粒C结合了点状和线性孔隙。结果显示,具有管状孔隙的颗粒A和C表现出高度各向异性膨胀,最大膨胀方向垂直于其管状孔隙取向,而具有点状孔隙的颗粒B在xy平面内呈现各向同性膨胀。
更重要的是,具有平面、异质纳米孔隙的硅颗粒会出现各向异性膨胀和早期开裂,而那些具有均匀、管状纳米孔隙的颗粒则表现出各向同性应变和结构完整性。研究人员建议将SOC上限设定在60-70%,这对应于线性膨胀后的拐点,有助于保持结构完整性和延长循环寿命。
三维微观结构的演化奥秘
Millennial Lithium
同步辐射X射线CT技术提供了电极三维微观结构演化的清晰视图。研究发现,碳粘结剂域(CBD)的膨胀对电极肿胀有着显著贡献。

石墨/μ-Si复合电极的三维微观结构演变
在石墨/μ-Si电极(17 wt.% Si)中,CBD膨胀(20 wt.%)显著促进了电极肿胀。X射线纳米CT显示,CBD增厚约25%,伴随着粗化和孔隙率损失,这与SEI形成和电解质吸收有关。这导致Li⁺通量减少和连接性下降。
突破性的双层电极设计
Millennial Lithium
基于这些发现,研究人员识别出石墨/μ-Si复合电极材料设计的五个关键挑战:
低CBD提升电池能量含量但限制硅利用率,而高CBD导致严重膨胀和孔隙率损失
高电极孔隙率促进电解质传输但牺牲能量和电接触
循环过程中硅损失使附近石墨过载,可能引发早期锂沉积
紧密的石墨包裹延迟硅锂化,降低其活性
硅膨胀阻碍电解质进入电极更深区域
为此,研究团队开发了一种异质双层石墨/μ-Si复合电极,其中顶层(隔膜侧)包含82 wt.%石墨 + 10 wt.%硅 + 8% CBD,底层包含20 wt.%石墨 + 60 wt.%硅 + 20 wt.% CBD,总硅含量为35 wt.%。

双层电极与参考均质石墨/μ-Si复合电极的物理和电化学性能比较
这种架构提供四个关键优势:空间分离减轻石墨过充并释放被包裹的硅;多孔顶层维持快速电解质传输;石墨丰富的顶层缓冲硅丰富底层的膨胀,同时压应力保持硅颗粒的结构和界面稳定性;底层高CBD含量确保循环过程中稳健的电接触。
实验结果显示,在锂金属纽扣电池配置中,这种设计相比传统电极提高了容量保持率(16个循环后72%对15%)。
这项研究揭示了颗粒内纳米孔隙、CBD演化、短程和长程三维结构对石墨/μ-Si复合电极锂化异质性和循环稳定性的影响。尽管技术成熟度较低,但在锂金属纽扣电池配置中测试的双层复合电极设计概念已显示出有希望的结果。
在考虑将这种电极设计作为实用大面积锂离子电池的可行候选方案之前,需要进一步优化至高电位锂离子电池架构并评估材料可扩展性。这项突破性研究为开发下一代高能量密度锂离子电池提供了重要的理论依据和实践指导。
通过精确调控纳米级孔隙结构和电极宏观架构,科学家们正在逐步攻克硅负极材料商业化应用的技术瓶颈,为电动汽车和储能领域带来新的发展机遇。
原文参考:Unravelling electro-chemo-mechanical processes in graphite/silicon composites for designing nanoporous and microstructured battery electrodes
*特别声明:本公众号所发布的原创及转载文章,仅用于学术分享和传递行业相关信息。未经授权,不得抄袭、篡改、引用、转载等侵犯本公众号相关权益的行为。内容仅供参考,如涉及版权问题,敬请联系,我们将在第一时间核实并处理。
-
动力型锂电池与普通锂电池的差别2016-01-07 7828
-
2016年十大锂电池技术突破2016-12-30 3983
-
石墨烯锂电池要问世啦!2017-01-16 5240
-
对于锂电池的开发将面临这样的挑战2017-01-17 4725
-
关于锂电池电极材料SEM测试、氩离子截面解剖电极片2017-07-07 9328
-
锂电池快充的奥秘2018-10-10 6489
-
Gocator三维智能传感器在锂电池缺陷检测的应用有哪些2020-07-31 3249
-
如何提高锂电池系统的能量密度?2021-03-11 2105
-
干掉锂电池,锌电欲别树一帜2021-04-06 2161
-
3.7V锂电池供电系统的设计资料分享2021-12-09 1520
-
TLM系列高能量锂电池2011-02-12 2135
-
硅材料有望成为高能量密度锂电池的负极材料优选2022-11-17 2907
-
锂电池和石墨烯电池哪个好?2023-08-22 14655
-
锂电池防爆要求 锂电池的防爆原理2024-01-10 4532
-
锂电池单体、锂电池组和锂电池包的区别2024-01-11 7704
全部0条评论

快来发表一下你的评论吧 !
