

MEGNet普适性图神经网络 精确预测分子和晶体性质
电子说
描述
近年来机器学习算法在包括自然语言处理,图像识别等诸多领域大放异彩。得益于如Materials Project1, QM92,3等材料数据库的不断完善和发展,机器学习正在被越来越多的应用在材料学领域的研究中。然而,由于研究目标的单一性,多数工作仍然局限于解决特定的晶体结构以及特定的材料性质预测问题。一种泛化的,普适性的机器学习模型仍然是材料学领域研究的重点目标。此研究是基于
基于DeepMind建立的图神经网络框架。

在材料学领域,对分子或晶体结构的特征描述需要满足平移,转动,镜面不变性,以及对整体结构特异信息的表征。常见的结构特征描述由于其局域性,缺乏对整体结构信息的表达,因而不具有普适性。图网络模型(graph-network)是一种基于图论的结构化模型,从理论上完美解决了这一问题。在图论中,图(graph)由若干给定的顶点(node)及连接顶点的边(edge)构成。运用到分子(或晶体)结构中,原子(atom)可以由顶点(node)描述,连接原子之间的化学键(bond)可以由边(edge)描述,从而可以把一个个分子或晶体结构看作一个个独立的“图”。基于此类模型的结构描述方法,科研工作者可以开发出针对任何材料结构或任何物理化学性质的普适性模型。尽管具有理论可行性,此类模型由于模型复杂度,材料数据量的限制,仍很少被应用在材料学领域中4,5。近日,UC San Diego 的 Shyue Ping Ong 课题组基于DeepMind建立的图神经网络框架6,开发了一套分子和晶体通用性质预测模型(MEGNet),在各项性质预测测试中达到了领先水平7。
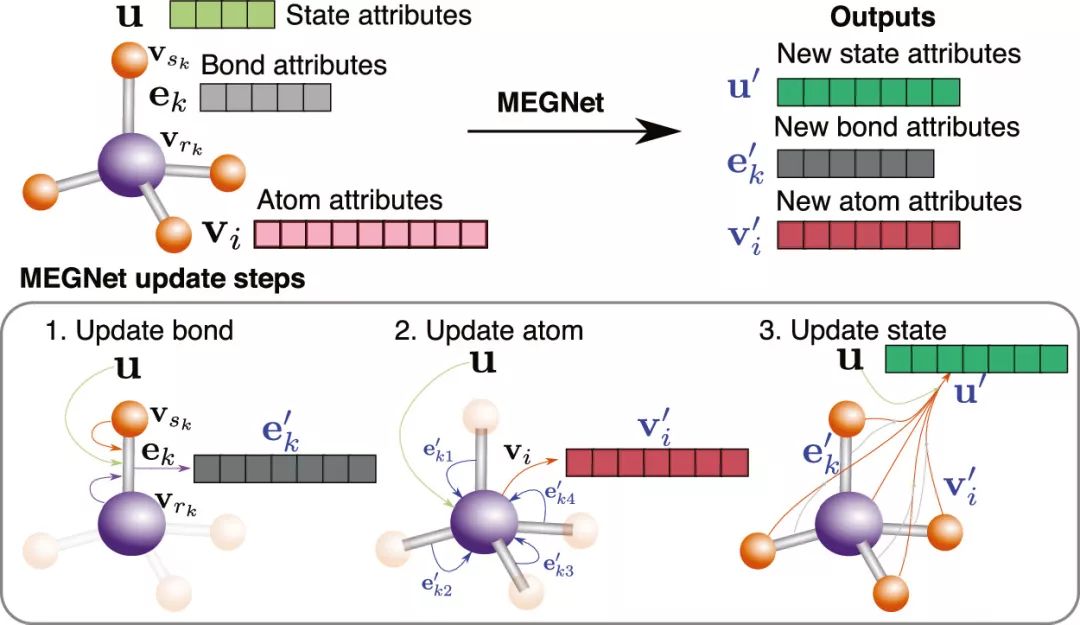
图 1. MEGNet 概述。每个分子/晶体结构由化学键信息,原子信息,和状态信息描述。每个结构描述输入模型后,依次更新,直到总的结构输出性质与DFT计算值接近。
图1中描述了该模型的工作模式: 每个结构可用三个向量表征,分别包含原子信息,化学键信息,和状态函数信息。在每一次模型训练迭代中,依次更新化学键向量,原子向量,和状态函数向量,得到新的结构表征向量,直到通过该表征输出的性质与DFT计算结果趋于一致。作者首先用QM9 分子数据集中超过130k数据作为训练集训练模型,并用得到的模型预测分子中的13项物理化学性质,在其中的11项中达到同类模型中的最优结果(表1)。更为先进的是,之前的工作对由状态参数关联的状态函数,如内能(U0, U),焓(H),和吉布斯自由能(G),采用的是分别训练模型进行预测的方法。
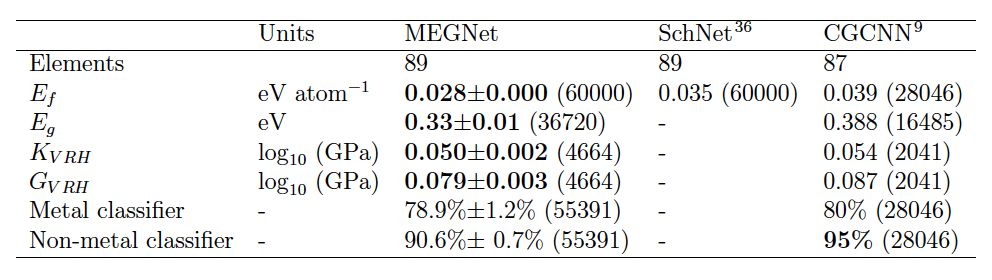
然而本工作中,作者采用加入状态参数作为输入的方法,可由单一模型同时预测U0, U, H和G,并保持与分别模型训练类似的准确度,大大提高了训练效率[YZ1] 。在针对晶体结构的应用中,作者用Materials Project数据库中超过69k数据作为训练集,针对生成能,能带带隙,体积模量和剪切模量进行了回归(Regression)分析,并用带隙值作为金属和非金属的判据进行分类(classification)分析。回归分析中的平均绝对误差(MAE)低于同类型模型SchNet4和CGCNN5(表2),金属和非金属分类分析中综合准确率达86.9%,ROC中AUC达到0.926,与此前最优模型CGCNN类似。
表1. 不同模型在QM9上预测13项性质的平均绝对误差(MAE)对比
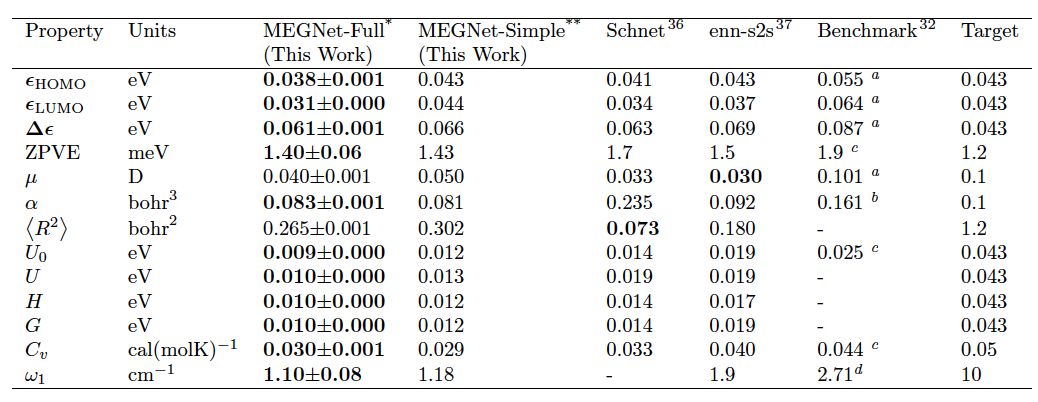
表2. MEGNet和其他基于图像模型
在Materials Project数据集的预测准确度对比
在对模型的深入分析中,作者发现,从最优模型中提取出的元素映射(embedding)与化学常识吻合。例如,将元素映射投影二维空间可发现,Eu和Yb与其他镧系元素距离较远,而与碱土金属更接近,这与化学经验相符。这样的分析一方面佐证了模型可以学习到可靠的化学信息,另一方面可将学习到的化学信息用于迁移学习,以大大降低训练新模型训练所需数据量。例如,在此例中,作者通过用~69k生成热的数据训练的模型提取的元素映射,用于预测带隙和弹性性质的模型训练,后者的数据量只有生成热的一半甚至十分之一。通过迁移学习的方法作者得到比直接训练更低的MAE和提高两倍的收敛速度。这为小数据量的性质的高效准确模型训练提供了可行的方案。
在模型的使用方面,用户可登陆http://megnet.crystals.ai,根据提示输入晶体结构编码或cif文件即可得到模型预测的性质。另外,文章所涉及的Python代码均已开源(https://github.com/materialsvirtuallab/megnet.git)。下面示例如何使用已有模型和训练新的模型。
1示例一: 使用分子模型

2示例二:使用晶体模型预测剪切模量

3示例三:训练新模型

-
bp神经网络模型怎么算预测值2024-07-03 1881
-
卷积神经网络和深度神经网络的优缺点 卷积神经网络和深度神经网络的区别2023-08-21 5244
-
卷积神经网络模型发展及应用2022-08-02 13342
-
如何构建神经网络?2021-07-12 1983
-
基于主动学习的半监督图神经网络模型来对分子性质进行预测方法2020-11-24 5275
-
【AI学习】第3篇--人工神经网络2020-11-05 4254
-
【案例分享】ART神经网络与SOM神经网络2019-07-21 3284
-
Keras之ML~P:基于Keras中建立的回归预测的神经网络模型2018-12-20 4438
-
BP神经网络的税收预测2018-02-27 1350
-
基于RBF神经网络的通信用户规模预测模型2017-11-22 1280
-
BP神经网络风速预测方法2017-11-10 1238
-
用matlab编程进行BP神经网络预测时如何确定最合适的,BP模型2014-02-08 3600
-
有提供编写神经网络预测程序服务的吗?2011-12-10 1741
全部0条评论

快来发表一下你的评论吧 !
